Introduction
Hemolytic uremic syndrome (HUS) is a prototype of thrombotic microangiopathy (TMA) and is characterized by the clinical triad of microangiopathic hemolytic anemia, thrombocytopenia, and acute kidney injury (AKI) [1]. The atypical form of HUS (aHUS) is distinguished from the typical form by the absence of a prior verotoxin-producing Escherichia coli infection. Notably, aHUS may be classified as a primary condition or may occur secondary to bone marrow transplantation, the use of specific drugs, pneumococcal or viral infections, and autoimmune diseases [1]. Primary aHUS is caused by abnormal activation of the alternative complement pathway, and various kinds of genetic mutations have been reported in patients with aHUS, including mutations in the genes encoding complement factor H (CFH), complement factor I, membrane cofactor protein (MCP/CD46), complement 3 (C3), complement factor B, and thrombomodulin, among others [2,3]. An acquired form of aHUS that primarily affects children aged 9ŌĆō13 years is observed in those who present with aHUS in association with anti-CFH autoantibodies (anti-CFHa-HUS) [4-6].
We report a case of a 13-year-old Lao girl with clinical features of aHUS, most likely anti-CFH-aHUS.
Case report
A 13-year-old previously healthy girl reported a weight gain of 3 kg (from 48 kg to 51 kg) over 3 weeks prior to presentation with gradually increasing edema. She visited a local hospital in Vientiane, Laos PDR and underwent laboratory testing that showed the following results: hemoglobin 6.2 g/dL, platelet count 76,000 cells/mm3, serum potassium 6.9 mEq/L, blood urea nitrogen (BUN) 205 mg/dL, serum creatinine 5.5 mg/dL, serum albumin 2.9 g/dL, and serum cholesterol 289 mg/dL. Urinalysis showed 4+ proteinuria and 4+ blood. She was clinically diagnosed with nephrotic syndrome and treated with oral prednisolone (60 mg/day), enalapril, and kalimate. She also received 20% albumin infusion (100 mL/day for 3 days), transfusion of 1 unit of packed red blood cells, and furosemide infusion (20 mg/day for 2 days). Despite this management, her condition did not improve, and she was transferred to ChildrenŌĆÖs Hospital, Vientiane, Laos PDR.
Upon admission, she appeared pale and edematous, although her consciousness was clear. She denied any preceding diarrheal or infectious diseases. Her family history was unremarkable without any history of renal and hematological diseases. She denied the use of any herbal medicine. Her body temperature was normal, her height and weight were higher than the 90th percentiles, systolic blood pressure varied between 120 and 140 mmHg, and her neurological status was E4V5M6 (using the Glasgow Coma Scale).
Her serum hemoglobin level was 6.7 g/dL, reticulocyte count was 3.3%, and platelet count was 103,000 cells/mm3. Serum sodium, potassium, and chloride levels were 134, 7.1, and 102 mEq/L, respectively. Serum uric acid level was 19 mg/dL. BUN and serum creatinine were 178 and 2.73 mg/dL, respectively, and serum albumin level was 3.2 g/dL. Her C3 level was 33 mg/dL (reference range 79ŌĆō1.52 mg/dL), and complement 4 level (C4) was 18 mg/dL (reference range 16ŌĆō38 mg/dL). Urinalysis revealed 4+ proteinuria and 4+ blood, and the spot urine protein to creatinine ratio was 1.47 mg/mg. Antinuclear antibody, anti-double stranded DNA antibody and anti-smooth muscle antibody were negative. Her anti-streptolysin O (ASO) titer was 200 IU/mL. Direct and indirect Coombs tests showed negative results. Hepatitis B and C virus markers were negative. Renal ultrasonography revealed that the kidneys were normal in size.
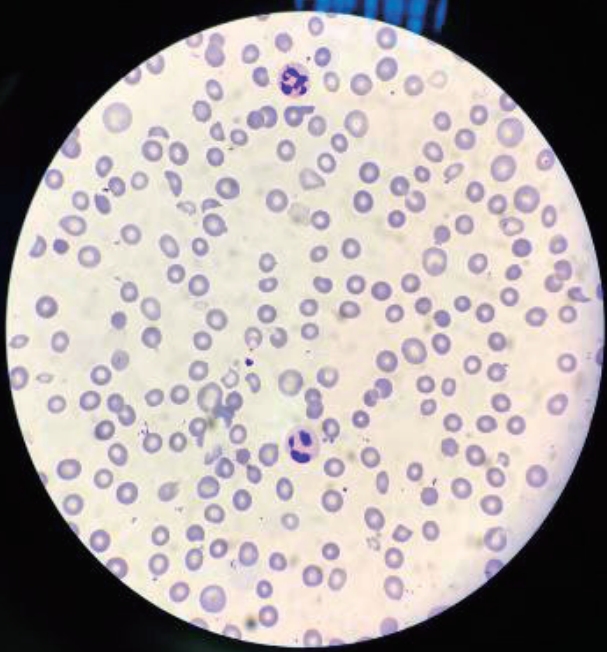
She was presumptively diagnosed with rapidly progressive glomerulonephritis (RPGN) secondary to acute poststreptococcal glomerulonephritis (APSGN) and treated with intravenous methylprednisolone (30 mg/kg/day) for 3 consecutive days followed by oral prednisolone (60 mg/day). Her serum creatinine decreased to 1.91 mg/dL; however, anemia and thrombocytopenia persisted. On the 7th day of hospitalization, she developed worsening renal function (BUN and serum creatinine levels were elevated to 143 mg/dL and 5.1 mg/dL, respectively) with gradually worsening oliguria. Peripheral blood smear examination showed abundant helmet cells and a reduced platelet count, suggesting TMA (Fig. 1). Based on the absence of preceding diarrheal illness, abnormal activation of the alternative complement pathway, and the age of disease onset, aHUS (possibly anti-CFH-aHUS) was considered the most likely diagnosis. Therefore, a second series of methylprednisolone pulse therapy (30 mg/kg/day for 3 consecutive days) concomitant with fresh frozen plasma (FFP) infusion therapy (10 mL/kg/day for 2 consecutive days) was instituted, which improved her renal function (serum creatinine 1.4 mg/dL), anemia (hemoglobin 9.5 g/dL), thrombocytopenia (349,000 platelets/mm3), proteinuria, and oliguria. However, a few days later, her symptoms again worsened with the development of infectious diarrhea and severe pneumonia. Despite the administration of large doses of antibiotics and repeated FFP infusions, she developed severe respiratory distress with bilateral pleural effusions (requiring mechanical ventilation), uncontrolled hypertension, and severe thrombocytopenia with petechiae and purpura. She developed generalized seizures and right-sided paralysis on the 40th day of hospitalization. Her parents decided to take her home, and she died at home. The serial laboratory findings of the patient were summarized in Table 1.
Discussion
The patientŌĆÖs initial clinical presentation including massive proteinuria, hematuria and azotemia indicated AKI due to glomerulonephritis. Furthermore, we suspected RPGN because of the rapid deterioration of renal function. This presentation combined with laboratory tests indicating borderline ASO titers and low C3 but normal C4 serum levels, led to a presumptive diagnosis of APSGN because this is the most common etiology in such patients. However, the occurrence of AKI, microangiopathic hemolytic anemia, and thrombocytopenia represented the clinical triad that fulfilled the diagnostic criteria for TMA. Among the various etiologies of TMA, anti-CFH-aHUS was the most likely condition in this patient, considering the absence of preceding diarrheal illness, abnormally activated alternative complement pathway, and her age at disease onset. A partial response to initial treatment with high-dose corticosteroids also suggested the diagnosis of anti-CFH-aHUS. Unfortunately, a kidney biopsy and laboratory tests to confirm the diagnosis were unavailable in our hospital and also in our country. Following the diagnosis of aHUS (possibly anti-CFH-aHUS), high-dose corticosteroid and plasma infusion therapy improved her general condition as well as her laboratory test results, including renal function and hematological profiles. These findings strongly suggested the diagnosis of anti-CFH-aHUS. However, the patient died of infectious complications.
Approximately 50% of patients with aHUS demonstrate genetic or autoimmune abnormalities causing dysregulation of the alternative complement pathway [7]. CFH is the most commonly affected gene, and patients with CFH mutations comprise 20ŌĆō30% of all patients with aHUS [7]. The presence of anti-CFH autoantibodies causes functional deficiency of CFH [4-6]. Anti-CFH-aHUS has been reported mainly in children aged 9ŌĆō13 years, although all age groups including infants and adults may be affected [6]. On the other hand, the onset age of pediatric aHUS in association of genetic mutations is primarily before the age of 2 years, and onset earlier than 3 months of age is characteristic of CFH and CIF mutation-associated aHUS [8]. The prevalence of anti-CFH-aHUS shows ethnic differences. However, prevalence in studies reporting exclusively pediatric cases (10ŌĆō56% of total cases with aHUS) [2,9-11] is higher than that in studies reporting both, pediatric and adult cases (5ŌĆō10%) [3,11,12]. A large cohort study that included patients with anti-CFH-aHUS [6] reported that abnormal activation of the alternative complement pathway (low serum C3 levels with normal C4 levels) was observed in 58% of the patients at disease onset, as was observed in our patient.
Interestingly, patients with anti-CFH-aHUS commonly present with severe abdominal pain and vomiting (84%) and diarrhea (53%) at disease onset [6]. Owing to these findings, anti-CFH-aHUS is often indistinguishable from the typical form of HUS. Additionally, extrarenal complications during the acute phase are more common than those in patients with aHUS in association with genetic mutations. The common complications are seizures in 23.5%, pancreatitis in 23.1%, and hepatitis in 50% of patients [6]. Our patient did not show any such extrarenal complications during the acute phase, but she developed seizures with hemiparesis later during the disease course.
The mainstay of treatment for anti-CFH-aHUS during the acute phase is early initiation of plasma exchange to remove the circulating autoantibodies and administration of corticosteroids and immunosuppressants (cyclophosphamide or rituximab) to reduce or prevent continued autoantibody production. This therapy is followed by maintenance immunosuppressant therapy (mycophenolate mofetil) [13]. Because plasmapheresis was unavailable in our hospital, the patient received repeated plasma transfusion instead. Although plasma infusion does not remove the circulating autoantibodies antibody, it can, at least partially, correct abnormal plasma complement profiles, which resulted from abnormal activation of the complement alternative pathway. Therefore, plasma infusion can be used as an alternative treatment for plasmapheresis when plasma exchange is unavailable or when vascular access for plasmapheresis is difficult. However, plasma infusion may result in or aggravate volume overload.
A few case reports have described that eculizumab is effective in these patients [14,15]; however, further studies are necessary to conclusively establish the role of eculizumab in patients with anti-CFH-aHUS. The long-term prognosis of anti-CFH-aHUS is similar to that of other forms of aHUS. Patients show a high frequency of relapse (59%), chronic kidney disease (39%), end-stage renal disease (27%), and death (9.5%) [6].
In conclusion, this is the first case report that describes aHUS in Laos. A 13-year-old girl presented with microangiopathic hemolytic anemia, thrombocytopenia, and AKI without preceding diarrheal illness. Low serum levels of C3 but normal levels of C4 indicated abnormal activation of the alternative complement pathway. Treatment with plasma infusion and high-dose corticosteroid therapy, which are the best possible treatment options available in Laos, led to improvement in her renal function and hematological profile. However, she died of infectious complications. Clinicians should suspect aHUS and evaluate further in patients presenting with azotemia, anemia and thrombocytopenia without preceding diarrheal disease, although it is very rare.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print