Introduction
Autosomal dominant polycystic kidney disease (ADPKD) is the most frequent hereditary renal disease which causes terminal chronic renal failure. It is characterized by bilateral multiple renal cysts produced by mutations to the genes PKD1 (85%) and PKD2 (15%).
Only 9 cases of unilateral ADPKD with agenesis of contralateral kidney have been reported all around the world , none of them were discovered before birth and none of them where described before having symptoms (Table 1).
We report the only case in the world of unilateral ADPKD with dysplasia of contralateral kidney in a 4-yr-old girl diagnosed intrauterine, since she was 20 weeks old and followed for 4 years until now.
Case report
A newborn female was studied in our hospital due to familiar history of ADPKD (father 38 years old had hypertension, grandmother and aunts had kidney transplantation in the last 10 years) and a renal dysplasia of the right kidney, diagnosed by prenatal ultrasound examination performed on the 20th week.
We proposed to have an amniocentesis to diagnosis ADPKD, but parents refused.
A new ultrasound examination done when newborn was 7 days old, revealed a 1mm cyst in the left kidney.
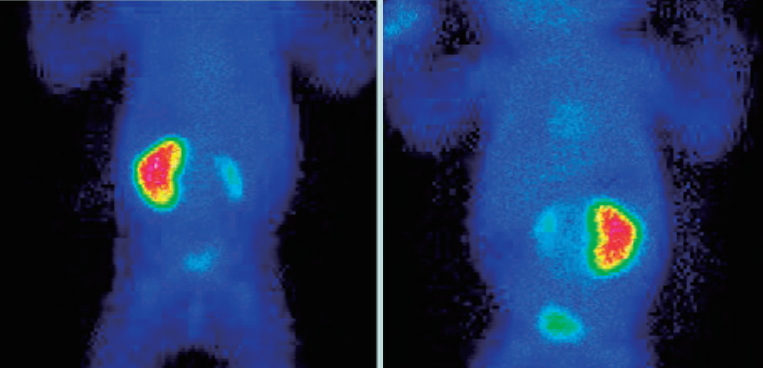
It was decided to do a scintigraphy 1 month after born, to describe real renal function of the patient. The scintigraphy showed a severe affected right kidney represented by a little sheet of functioning tissue, and a tubular mass of the 10%. Left kidney was morphologically normal and his function was above 90% (Fig. 1).
Newborn did not present any signs or symptoms of renal disease after birth, she was born with a normal birth weight and physical examination did not show any alteration. She did not present arterial hypertension. She presented a normal renal function, serum creatinine level 0ŌĆÖ5 mg/dL, sodium 132 mEq/L, and potassium 3 mEq/L. Urinalysis didn┬┤t show hematuria, or proteinuria. She did not present any urinary infection in her first years.
After she was born her family decided to make a genetic study of the whole family, which shows a PKD1 mutation in three generations, including our patient.
At the moment the 4 yr-old child does not present any symptom, last ultrasound examination shows an increased 2 mm cyst in the left kidney with no other alterations. Renal function, blood pressure and analytic parameters are absolutely normal.
Discussion
ADPKD, with an incidence as high as 1:400, affects more than 13 million individuals worldwide, and accounts for 7ŌĆō10% of end stage kidney disease (ESKD) in adults [1].
Over the past 20 years, it has become clear that ADPKD, previously known as ŌĆ£adult polycystic kidney diseaseŌĆØ is an important clinical entity in the pediatric population which affect all age groups children.
Indeed, 60% of children younger than 5 years of age, and 75 to 80% of children 5 to 18 years of age with a PKD-1 mutation have renal cysts detectable by ultrasound studios [2,3].
Renal cysts in children with ADPKD have been associated with a wide clinical spectra, totally asymptomatic patients to those who present massive renal enlargement, hypertension, oliguria, and pulmonary hypoplasia.
We described the first case in the world of unilateral ADPKD diagnosed in the pregnancy. There are 8 cases of unilateral ADPKD described all around the world, but none of them have been described or followed since born. (Table 1).
Unilateral renal agenesis occurs in approximately 1 in 1000 births, and is generally a sporadic anomaly. Except for ectopia, malrotation or hydronephrosis, abnormalities of the contralateral kidney are infrequent. Unilateral renal agenesis is usually asymptomatic.
At the moment, it doesnŌĆÖt exist any verified information about unilateral ADPKD evolution, prognosis, or treatment, because cases are not being reported in a sufficient quantity, but, as much as prenatal diagnosis has being improved, unilateral ADPKD cases could be published and studied, and that would be a great tool in the future to investigate and advance in one of the most important genetic renal illness worldwide.
We remark the importance of report this case because there are not clear rates of morbidity or mortality of these patients. It is necessary, to have a real information about prognosis in unilateral ADPKD with contralateral agenesis.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print