Pathogenesis of Minimal Change Nephrotic Syndrome: A Review of the Underlying Molecular Mechanisms
Article information

Abstract
Nephrotic syndrome (NS) is the most common glomerular disorder in childhood, and a vast majority of cases are idiopathic. The precise cause of this common childhood disease is not fully elucidated despite significant advancements in our understanding of podocyte biology. Idiopathic NS has been considered “a disorder of T-cell function” mediated by a circulating factor that alters podocyte function resulting in massive proteinuria since the last four decades. Several circulatory factors released from T-cells are considered to be involved in pathophysiology of NS; however, a single presumptive factor has not been defined yet. Extended evidence obtained by advances in the pathobiology of podocytes has implicated podocytes as critical regulator of glomerular protein filtration and podocytopathy. The candidate molecules as pathological mediators of steroid-dependent NS are CD80 (also known as B7-1), hemopexin, and angiopoietin-like 4. The “two-hit” hypothesis proposes that the expression of CD80 on podocytes and ineffective inhibition of podocyte CD80 due to regulatory T-cell dysfunction or impaired autoregulation by podocytes results in NS. Recent studies suggest that not only T cells but also other immune cells and podocytes are involved in the pathogenesis of MCNS.
Introduction
Nephrotic syndrome (NS) is the most common glomerular disease in children. The estimated incidence of NS is one to sixteen cases per 100,000 children, varying by ethnicity and region [1]. The incidence among certain ethnic populations, specifically South Asian and African, is higher, which indicates that environmental and/or genetic factors affect the onset of the disease [2]. NS is characterized by the triad of heavy proteinuria, hypoalbuminemia (≤2.5 g/dL), and generalized edema [1]. Excessive leakage of serum proteins into urine results in a hypercoagulable state, increased rate of infection, and dysregulation of water balance [3]. Fortunately, 80% of children with NS respond to corticosteroid therapy and long-term kidney outcome is excellent, with risk of progression to chronic kidney disease estimated to be less than 5% in children with steroid-sensitive NS, at 10 years after diagnosis [4].
NS is generally divided into primary or idiopathic, secondary, and congenital, with idiopathic (INS) being the most common form observed in 90% of children. Glomerular lesions associated with INS include multiple histologic types, such as minimal change NS (MCNS), focal segmental glomerulosclerosis, membranoproliferative glomerulonephritis, and membranous nephropathy [1,5]. All these types of morphology share a common feature with podocyte foot process effacement and structural disorganization of the glomerular filtration barrier leading to proteinuria. These ultrastructural abnormalities typically resolve with corticosteroid-induced clinical remissions [5]. However, the mechanisms of this acquired and reversible abnormality in glomerular ultrastructure and filtration barrier permeability are incompletely understood.
Shalhoub, in 1974, proposed that the increased glomerular permeability to plasma protein in MCNS is due to a circulating factor released by T-cells; however, until date the search for the circulating factor has been unsuccessful. In addition, advances in the knowledge of podocyte pathobiology in the mechanism underlying proteinuria have led to NS being recognized as a podocytopathy. Based on this knowledge, several hypotheses have been proposed to explain the role of podocytes and related molecules in the mechanism underlying proteinuria in INS. Although the pathophysiology of NS has not been completely explained until now, it is considered a complex multifactorial disease with an immunological component. In this review, we discuss recent research findings in the pathogenesis of MCNS.
Pathogenesis of MCNS
Podocyte foot process effacement is the ultrastructural hallmark in MCNS, however, the pathogenesis leading to podocyte effacement is not clear. Systemic factors, immune mediated or circulating, and local factors can contribute to podocyte effacement, but there is no single unifying hypothesis (Fig 1).
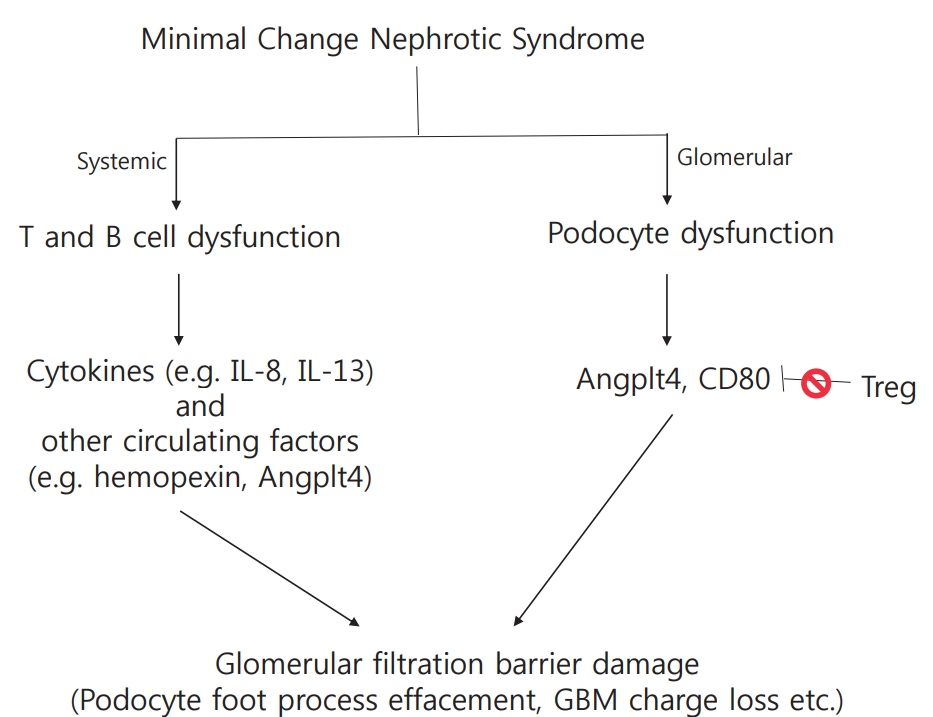
Pathogenetic pathways leading to minimal change nephrotic syndrome. Angplt4, angiopoitin-like 4; Treg, regulatory T-cell;GBM, glomerular basement membrane.
Immune dysregulation
As immunosuppression with corticosteroids is the mainstay of NS therapy, it is logical to suspect immune dysregulation as a pathogenic factor in disease development. In 1974, Shalhoub postulated that MCNS is a disorder of T-cell function, resulting in increased plasma levels of lymphocyte-derived permeability factor [6]. This hypothesis was based on the following clinical observations: [1] remission is commonly accompanied by measles infection whereby cell-mediated immunity is suppressed; [2] MCNS is associated with Hodgkin’s disease, which is a known T-cell disorder; [3] patients show good response to corticosteroids and cyclophosphamide, which are inhibitors of T-cell function; [4] humoral component deposition (immunoglobulin and components) is absent in glomeruli, which is unlike that in other glomerular disorders. Therefore, the massive proteinuria and hypoalbuminemia that characterize NS were thought to result from increased glomerular capillary wall permeability due to T-cell activation triggered by several stimuli, such as viral infection or allergens.
1. Role of cytokines
Investigators have made attempts, based on Shalhoub’s hypothesis, to identify the circulating factors released from T-cells that increase glomerular permeability to serum proteins, and some studies have confirmed that capillary permeability factor is detectable in patients with NS [7,8]. Of the various factors presumed to increase glomerular permeability to serum proteins, the most likely pathogenic factors are considered to be cytokines, which are small proteins secreted by the cells of both the innate and adaptive immune systems that transfer information within the immune system [9,10]. Patients who suffered relapses were found to have elevated serum or urine levels of various cytokines, including interleukin (IL)-2 [11,12], soluble IL-2 receptor [11,13], interferon-gamma [11,12], IL-8 [14-16], IL-13 [17-19], tumor necrosis factor-α [20], and vascular endothelial growth factor [21]. Of the many known cytokines, IL-8 and IL-13 in particular have been proposed to be most likely to be circulating factors. IL-8 may play a role in proteinuria by affecting the metabolism of glomerular basement membrane (GBM) components [15]. Additionally, urinary IL-8 levels were higher in patients who suffered relapses and had a positive correlation with the degree of proteinuria [16]. However, rat podocytes incubated in vitro with high concentrations of human IL-8 showed no difference in GBM permeability [22]. IL-13 has been revealed to stimulate intracellular podocyte protein trafficking and proteolysis in vitro [17]. IL-13-transfected rats developed severe proteinuria and showed MCNSlike nephropathy [18]. Increased IL-13 also induced the overexpression in podocytes of CD80, recently identified as a possible molecular mechanism underlying proteinuria in NS [23]. However, not all patient with MCNS present serum IL-13 elevation during relapse, and serum IL-13 is also known to be increased in clinical conditions not associated with proteinuria, such as allergy including asthma and atopy [24,25]. Studies conducted over the last 40 years have reported conflicting results regarding the role of cytokines in MCNS.
2. Role of regulatory T-cells (Tregs) and B-cells
Historically, MCNS has been considered a T-cell disease; however, advances in the study of basic immunology have contributed to a more articulated understanding of its pathogenesis taking into account Tregs and B-cells. Normally, cytokine release by T-cells is transient owing to the activation of Tregs that interact with T effector cells to suppress cytokine production. Tregs have been suggested to constitute a second step in an MCNS cascade, of which the first remains unclear [26,27]. The induction of Treg led to a marked reduction in proteinuria in animal models, and most patients with MCNS showed decreased levels of Treg [28,29]. Shimada et al. reported that abnormal censoring of podocyte CD80 expression could underlie Treg dysfunction or impaired autoregulation by podocytes. Treg dysfunction could lead to transient massive proteinuria becoming persistent, following which podocyte injury, and eventually, MCNS, occur [27]. Unlike role of T-cells in MCNS, which has been extensively studied, the role of B-cells is currently not well understood. Clinical trials have been conducted that demonstrated MCNS remission after B-cell depletion using the anti-CD20 monoclonal antibody rituximab [30,31]. The recent successful use of anti-CD20 monoclonal antibodies for the treatment of steroid sensitive NS raises the possibility of B-cells either influencing T-cells or themselves being primary players in NS. CD80 is expressed by both activated B- and T- cells, and increased nitric oxide production by B-cells observed in NS patients with relapse further supports the possibility of B-cell involvement [10,32]. Altogether, these results revealed that not only T-cells, but also B-cells or B-cell products might be implicated in the causal mechanism of MCNS via the abnormal regulation of T-cell function by circulating B-cells or by communication between B- and T-cells [33]. However, information on the role of B-cells is currently limited.
Systemic circulating factors
1. Hemopexin
Hemopexin is an abundant plasma protein that effectively scavenges heme. It has been proposed that various isoforms of hemopexin exist [10]. Under normal conditions, circulating hemopexin is inactive, but under certain conditions, it becomes activated as a serine protease [34]. The active isoform of hemopexin has been reported to be implicated in the pathogenesis of MCNS. Infusion of human plasma hemopexin in rats induces reversible proteinuria accompanied by podocyte foot process effacement and loss of the negative charge of the GBM [35]. Furthermore, hemopexin induces nephrin-dependent cytoskeletal rearrangement in podocytes and affects permeability of the glomerular filtration barrier by reduction in glycocalyx. The effects of hemopexin were inhibited by pretreatment with normal human plasma and serine protease inhibitors [36]. Hemopexin is suspected to be a circulating factor causing MCNS, although the mechanism of hemopexin activation and the inhibitory factors that activate hemopexin under normal conditions is unclear.
2. Angiopoietin-like 4 (Angptl4)
Angptl4 is a glycoprotein secreted mainly in the liver and adipose tissue. Angptl4 is minimally expressed in normal glomeruli, but it is highly upregulated in the serum and podocytes in experimental models of MCNS and in the human disease [10,37,38]. There are two isoforms of podocyte-secreted Angptl4: the hyposialylated form secreted by podocytes (podocyte-secreted form), and the sialylated form secreted by skeletal muscles, heart, and adipose tissue (circulating form) [39]. Podocyte-secreted Angtpl4 is glucocorticoid sensitive and has been proposed to be a mediator of proteinuria. It was observed that its upregulation induced massive proteinuria, loss of negative charge in the GBM, and foot process effacement in vivo models of NS, and that its conversion into sialylated Angptl4 [37]. In addition, circulating Angptl4 was secreted in response to an elevated plasma free fatty acid to albumin ratio when proteinuria reached the nephrotic range, subsequently resulting in hypertriglyceridemia [38]. This indicates that Angptl4 can be developed as a biomarker of MCNS in feature studies.
Co-stimulatory molecule CD80
CD80, also known as B7-1, is a transmembrane molecule present on the surface of both antigen presenting cells and activated B-cells, and acts as a co-stimulatory signal for T-cell activation [10]. CD80, present on the surface of antigen presenting cells, binds CD28 on effector T-cells or cytotoxic T lymphocyte-associated protein 4 (CTLA4) in regulator T-cells, determining T-cell activation (CD28) or inhibition (CTLA4) [40]. In 2004, Reiser et al. reported that under certain conditions, podocytes can express CD80, and its expression results in the development of a proteinuric condition [23]. Proteinuria was not induced in CD80 knockout mice by lipopolysaccharides administration, but it was induced in SCID mice, which are deficient in T- and B-cell functions, showing that CD80 plays a key role independent of T- and B-cells [23]. Increased CD80 levels in urine are observed in patients with MCNS with relapse compared to those in remission and with other glomerular diseases (lupus, focal segmental glomerulosclerosis) [41,42]. A recent study has reported that high urinary CD80 excretion might be a biomarker for steroid responsiveness and a predictor for good prognosis in NS [43]. In addition, polyinosinic:polycytidylic acid (polyI:C), a ligand of Toll-like receptor 3 which mimics viral infection, promotes podocyte CD80 expression [44]. This offers a possible reasoning to explain the frequent relapse of MCNS after upper respiratory virus infection. Because PolyI:C induces only transient proteinuria, impaired regulatory mechanisms after CD80 induction were postulated as a second hit cause of MCNS [27]. Suppression of CD80 expression could be a novel therapeutic strategy for MCNS; however, more evidence is required to support this idea.
Conclusions
The pathophysiology of NS is still far from being fully explained, although recent advances in podocyte biology have provided novel insights into the possibility of NS being a podocytopathy. MCNS is regarded as a multifactorial disease. It is hypothesized that MCNS is a podocytopathy and that CD80 or other circulatory factors are the triggers for proteinuria. Recent and future research will lead to new therapeutic targets in MCNS.
Notes
Conflict of interest
This study was funded by Individual Basic Science & Engineering Research Program through the Ministry of Education of the Republic of Korea and National Research Foundation of Korea (NRF-2016R1D1A1B03933207)