Comprehensive review of membranoproliferative glomerulonephritis: spotlighting the latest advances in revised classification and treatment
Article information

Abstract
Membranoproliferative glomerulonephritis (MPGN) is a complex group of renal diseases characterized by a specific pattern of glomerular injury that includes thickening of the capillary wall and mesangial expansion, leading to a heterogeneous group of conditions. This review article offers a comprehensive overview of MPGN, its new classification, pathophysiology, diagnostic evaluation, and management options.
Introduction
Membranoproliferative glomerulonephritis (MPGN) is not a disease but a pattern of glomerular injuries characterized by thickening of the capillary wall (remodeling with formation of double contours) and mesangial expansion due to increased matrix deposition and hypercellularity observed on light microscopy [1-3]. MPGN most commonly presents in pediatric populations but can occur at any age [1]. There are no specific symptoms that uniquely represent MPGN; patients can present with various forms, including asymptomatic hematuria and proteinuria, acute nephritic syndrome, nephrotic syndrome, renal insufficiency, and even rapidly progressive glomerulonephritis. Hypertension and hypocomplementemia (complement components; C3 and/or C4) are often but not always present. These diverse clinical presentations result from differences in the underlying pathogenesis [1,2,4]. The condition can lead to progressive kidney damage and may ultimately result in renal failure if left untreated. The treatment depends on the etiology [5]. In this review, we explore the key aspects of MPGN to provide a comprehensive understanding of this complex renal disorder using a new classification.
Classically, MPGN was divided into three types based on the location of deposits found on examination by electron microscopy: primary (idiopathic) MPGN types I, II or III and secondary MPGN. The most common form, MPGN I, is characterized by subendothelial and mesangial electron deposits, while MPGN III exhibits both subepithelial and subendothelial electron deposits. MPGN II is also known as dense deposit disease (DDD) due to its electron-dense intramembranous deposits. However, this prior classification was not based on disease pathogenesis, resulting in pathogenetic heterogeneity.
New classification and pathophysiology
In most cases, MPGN results from deposition of immunoglobulins and/or complement in the glomerular mesangium and capillary wall when there are an increased level of circulating immune complexes and/or dysregulation of the alternative complement pathway. Advances in understanding the underlying disease have led to a new pathobiology-based classification of MPGN based on immunofluorescence findings. The MPGN pattern of injury is now classified into three groups (Fig. 1) [1,2,4].

Immunoglobulin/immune complex-mediated (immunoglobulin subgroup with or without a complement subgroup)
This can be present in two forms: immune complex-mediated glomerulonephritis (ICGN) with an MPGN pattern and glomerulonephritis with monoclonal immunoglobulin deposits.
ICGN with an MPGN pattern
This subtype arises from deposition of immune complexes within the glomeruli, leading to a distinctive MPGN pattern. It commonly occurs in association with various underlying conditions, including viral infections such as hepatitis C and hepatitis B; bacterial infections such as endocarditis and infected ventriculo-atrial shunts; and protozoa/other infections such as malaria, schistosomiasis, mycoplasma, leishmaniasis, filariasis, and histoplasmosis. The subtype can also be linked to autoimmune diseases such as systemic lupus erythematosus, Sjögren’s syndrome, rheumatoid arthritis, and mixed connective tissue disease. In some cases, it may manifest idiopathically when none of these specific conditions are evident. Diagnosis of this condition begins with a comprehensive evaluation aimed at uncovering the underlying disease trigger. However, when extensive assessment fails to reveal an underlying cause, it becomes necessary to explore the possibility of complement dysregulation [6,7]. This involves a systematic approach, encompassing the following steps: (1) Functional assay: tests for CH50, AP50 (complement alternate pathway activation 50%), and factor H function; (2) Quantification of complement component regulators: comprehensive assessment of C3, C4, factor I, factor H, factor B, and properdin levels; (3) Measurement of complement activation: evaluation of complement activation markers such as complement component 3d, activated factor B, and soluble membrane attack complex; (4) Autoantibody testing: screening for autoantibodies against factor H, factor B, and nephritic factors (C3, C4, C5); (5) Genetic testing: genetic analysis involving C3, complement factor H, complement factor I, complement factor B, and complement factor H-related proteins 1–5 through multiplex ligation-dependent probe amplification; (6) Plasma cell disorder assessment: examination of serum-free light chains, serum and urine electrophoresis, and immunofixation; (7) Immunofluorescence studies on kidney biopsy specimens: comprehensive immunofluorescence analysis of kidney biopsy specimens to detect the presence of immunoglobulin A, immunoglobulin G, and immunoglobulin M, C1q, C3, fibrinogen, kappa, lambda, and C4d. Notably, C4d typically exhibits a distinctive pattern characterized by bright staining, while immunoglobulin levels are often zero or minimal [8].
Glomerulonephritis with monoclonal immunoglobulin deposits
This subtype is observed in patients with monoclonal gammopathies and infrequently in patients with overt hematologic diseases such as multiple myeloma, Waldenström macroglobulinemia, or B-cell lymphoma. Kidney injury results from direct glomerular deposition of monoclonal immunoglobulins. Diagnosis requires an evaluation of the presence of a hematologic malignancy. Tests such as serum and urine protein electrophoresis, immunofixation, and serum-free light chain levels are necessary, and consultation with hematologists should be carried out as needed [9].
Complement-mediated (complement-dominant subgroup)
This can be divided into C3/C4 glomerulopathy (C3G/C4G) and then further divided into DDD and C3/C4 glomerulonephritis (C3GN/C4GN). An evaluation of the etiology requires consideration of the alternative pathway of the complement [10,11].
C3/C4 DDD
C3/C4 DDD is defined by highly electron-dense osmophilic, predominantly intramembranous deposits.
C3 glomerulonephritis
C3GN shows C3 deposition at least two orders of magnitude greater than any other immune reactant. The alternative complement pathway is presumed to be the underlying mechanism. Other C3-dominant glomerular diseases, such as infection-related glomerulonephritis, must be excluded. Hypocomplementemia is present in only about 50% of cases [12,13].
C4 glomerulonephritis
C4GN is characterized by bright C4d staining with minimal or no deposition of C3 or immunoglobulin [14].
Membranoproliferative pattern without immune complexes or complement (immunofluorescence-negative subgroup)
An absence or traces of immunoglobulin or complement suggests thrombotic microangiopathy [2]. The change in classification from electron-microscopic to immunofluorescence-microscopic criteria has resulted in limited application of evidence from previous controlled trials to guide high-quality management decisions. Consequently, recommending appropriate management approaches for various diseases with MPGN injury patterns has become challenging [2,15,16].
Management
The reclassification of MPGN has created a significant knowledge gap in guiding the prescription of the most effective treatment modalities within the framework of the new MPGN classifications. Historically, the study by Tarshish et al. [17] underscored the positive impact of corticosteroid treatment on the prognosis of idiopathic MPGN in pediatric patients. In contrast, a recent study led by Kirpalani et al. [18] reclassified pediatric MPGN cases using an innovative taxonomy and demonstrated the effectiveness of corticosteroid treatment in improving estimated glomerular filtration rate (eGFR) in C3G, although such benefits were not observed in ICGN cases. Therefore, the previous treatment recommendations may not be directly applicable to the new classification, underscoring the necessity for further well-controlled studies to establish guidelines for the optimal management of pediatric MPGN. While the following management proposals are for clinical decision-making, knowing that they are based on low-quality evidence is crucial.
Immune complex-mediated glomerulonephritis
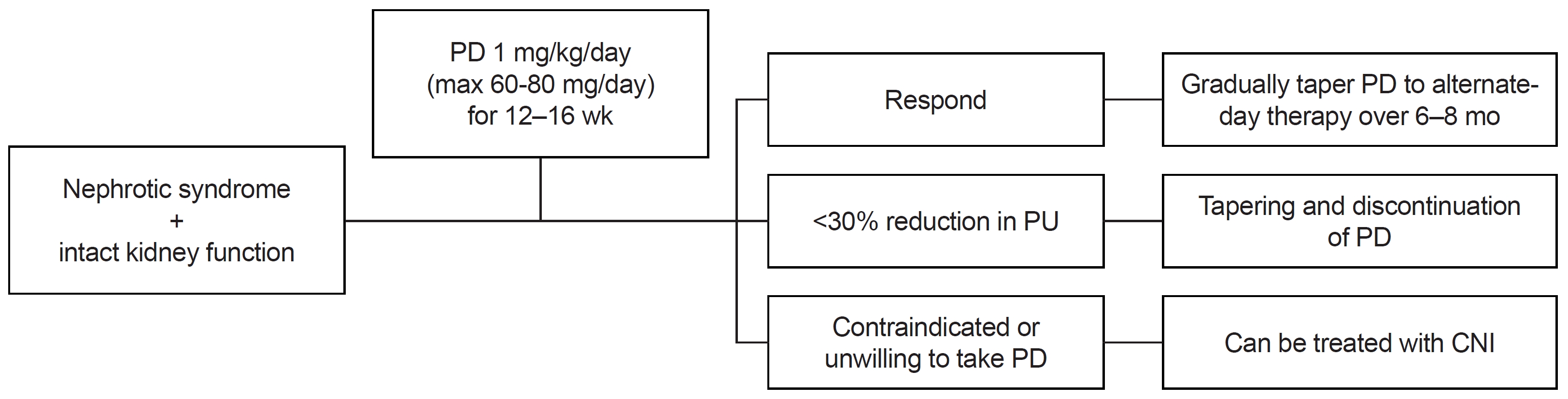
Assessing the underlying disease is the primary basis for treatment. Once the cause of ICGN is identified, treatment should focus on the underlying pathological process [19]. However, in cases where the cause remains unknown or is classified as idiopathic, it is advisable to consider the following steps. For those with nonnephrotic-range proteinuria and normal renal function, renin-angiotensin system inhibition may be a suitable option. In pediatric patients, the threshold for initiating immunosuppression may be lower than in adults, and mycophenolate mofetil could be considered as a glucocorticoid-sparing agent. Conversely, in cases of nephrotic syndrome with normal or near-normal renal function, glucocorticoid therapy should be considered (detailed management suggestions are provided in Fig. 2). Additionally, when abnormal kidney function is observed, particularly in the absence of crescentic involvement but with an active sediment, a comprehensive approach involving both glucocorticoids and immunosuppressive therapy coupled with supportive care is warranted. Detailed management recommendations for this scenario can be found in Fig. 3. Rapidly progressive crescentic ICGN may necessitate a more aggressive approach, involving high-dose glucocorticoids (typically 1–3 g of methylprednisolone) and cyclophosphamide. Finally, if renal function deteriorates to an eGFR of less than 30 mL/min/1.73 m², a focus on supportive care is crucial, along with an evaluation of kidney transplant options. Each treatment approach should be carefully considered in the context of the patient's clinical status and response to therapy to achieve the best possible outcomes [2].

Abnormal kidney function (without crescentic involvement) and active urine sediment with or without nephrotic-range proteinuria treatment suggestion [2]. PD, prednisolone; PU, proteinuria; MMF, mycophenolate mofetil.
C3 glomerulopathy
At present, the optimal management strategy remains poorly defined. For cases of moderate-to-severe disease characterized by moderate-to-marked proliferation on biopsy and proteinuria exceeding 2 g/day, consideration of immunosuppression is warranted [20]. Additionally, in patients presenting with both proteinuria >1 g/day and hematuria or experiencing persistent kidney function decline over a minimum of 6 months, the initial treatment should involve a combination of mycophenolate mofetil and glucocorticoids. If this initial approach is ineffective, it may be prudent to explore the potential benefits of eculizumab. If a patient does not respond favorably to the aforementioned management strategies, participation in clinical trials, where available, should be considered [6,12,13,20-24].
Prognosis
The renal and patient outcomes in adult MPGN patients are known to be poor [12,13,25]. A similar result was found in Korean adults by Lee et al. [26]. They found the MPGN had the worst renal and patient outcomes among primary glomerulonephritis. The prognosis of MPGN in Korean children is yet to be known. Pediatric patients diagnosed with ICGN and C3G often experience a more favorable clinical course compared with adult patients, marked by well-preserved renal function [18]. Progression to advanced chronic kidney disease is rare in children. However, C3G tends to carry a more unfavorable renal prognosis when contrasted with ICGN. In cases of C3G, early steroid intervention may be beneficial [18]. Additionally, hypertension is an independent risk factor for poor renal outcomes in adult MPGN [25]. Like adults, pediatric patients with hypertension showed lower eGFR compared to normotensive [18]. Therefore, hypertension control may be beneficial for pediatric MPGN patients. However, current pathologic criteria alone may prove insufficient for accurate prognosis in children presenting with ICGN and C3G, and further research is warranted [18].
Conclusion
MPGN is a pattern of glomerular injury, not a disease, and is a complex renal condition that requires a thorough evaluation to reveal the underlying causes and indicate effective management options. Early diagnosis and appropriate treatments are crucial for improving the prognosis and quality of life of individuals living with MPGN. As our understanding of the disease continues to evolve with the changed classification based on pathophysiology, new treatment options and approaches offer hope for better outcomes for affected individuals.
Notes
Conflicts of interest
Jeong Yeon Kim is an editorial board member of the journal but was not involved in the peer reviewer selection, evaluation, or decision process of this article. No other potential conflicts of interest relevant to this article were reported.
Funding
None.
Author contributions
All the work was done by JYK.