Desmopressin responding female nephrogenic diabetes insipidus: a case report
Article information

Abstract
Nephrogenic diabetes insipidus, decreased ability to concentrate urine, with production of large amounts of urine, is caused by the refractory response of renal tubules to the action of antidiuretic hormone. This rare disorder, known as X-linked nephrogenic diabetes insipidus, is caused by a mutation in the AVPR2 gene. Because it is hereditary, most patients are male. This report highlights a case of nephrogenic diabetes insipidus in a 3-year 5-month-old female; upon presentation to the hospital, her symptoms included frequent urination and consumption of a significant amount of water, which had begun 2 years ago. The results of blood tests showed increased levels of serum antidiuretic hormone, and sellar magnetic resonance imaging showed no abnormality. The results of the water restriction test and the desmopressin administration test confirmed the diagnosis of nephrogenic diabetes insipidus showing a partial response to desmopressin. The results of genetic testing indicated the presence of an AVPR2 mutation, a heterozygous missense mutation (p.Val88Met), suggesting inheritance of X-linked nephrogenic diabetes insipidus. This report describes a significant case of symptomatic X-linked nephrogenic diabetes insipidus in a female patient who showed a partial response to desmopressin.
Introduction
Nephrogenic diabetes insipidus (NDI), a decrease in the ability to concentrate urine, results from refractory to the action of antidiuretic hormone (ADH) [1]. Most patients with NDI harbor the X-linked recessive mode of inheritance (XL-NDI) resulting from loss-of-function mutations in the AVPR2 gene, encoding arginine vasopressin V2 receptor. Therefore, most of severe effects are observed in male XL-NDI patients with a hemizygous AVPR2 mutation, while cases involving less symptomatic female carriers, resulting from skewed X inactivation, have also been reported [2]. We report on a case of a 3-year 5-month-old female patient with a heterozygous AVPR2 mutation. The patient was diagnosed with polyuria, and showed a partial urinary concentrating response to treatment with desmopressin (1-desamino-8-D-arginine vasopressin).
Case report
A 3-year 5-month-old female presented with symptoms that included prolonged fever and polydipsia. The patient’s height was 97 cm (25th–50th percentile), weight; 18.1 kg (95th–97th percentile), water intake; 1.5–1.8 L/day, urination frequency; 7 times/day, voiding amounts; 1,000 mL/day (55 mL/kg/day). The patient's history included recurrent fevers that began at 7 months of age, however, an evaluation conducted in the hospital found no source of infection. Thus, she was transferred from hospital to hospital. She presented to our hospital at the age of 1 year and 6 months for evaluation of her persistent fever; her mother said that she had been consuming a significant amount of water. Testing of the urinary system was performed. A kidney and bladder ultrasound and renal dimercaptosuccinic acid scan showed no abnormal findings, however, results from repeated testing showed elevated levels of serum ADH, as shown in Fig. 1. Blood glucose and electrolytes including serum sodium were in the normal range. The symptoms of polydipsia were not severe, therefore, the patient’s ADH level was assessed every 6 months. The results of testing showed consistently high levels of ADH, and genetic testing indicated that she carried a heterozygous Val88Met mutation in the AVPR2 gene; and an additional arginine vasopressin (AVP) gene test did not detect the presence of a pathogenic mutation on the AVP gene (Fig. 2). No abnormal finding was detected on sellar X-ray and magnetic resonance imaging performed in the hospital. As shown in Table 1, the results of the water restriction test showed an increase in urine osmolality from 115 to 261 mOsm/kg at 6 hours post-restriction; the urine osmolality had doubled to 579 mOsm/kg at 2 hours after administration of desmopressin (Minirin tab 0.2 mg, per oral). The results of the water restriction test showed that urine osmolality was higher than plasma osmolarity in response to desmopressin, however, based on an increase to less than 800 mOsm/kg and the result of genetic testing, the patient was diagnosed with partial NDI. Genetic analysis was not performed on the parents because there was no symptoms of NDI and no plan for another child.
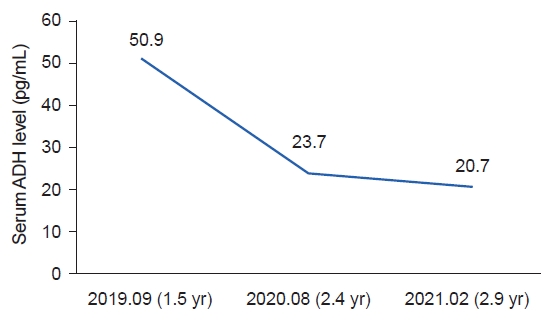
Serial test results of serum antidiuretic hormone (ADH) levels with the decreasing tendency (ADH reference range : 0-14.0 pg/mL).
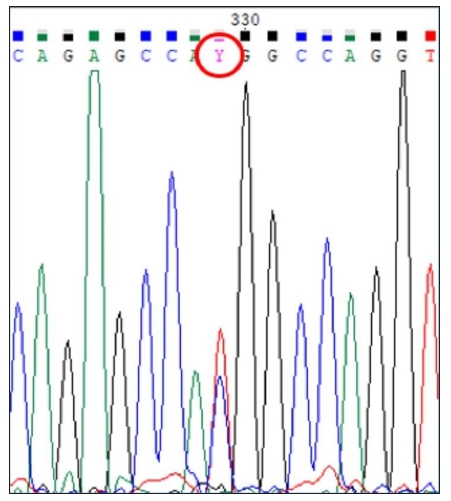
Result of DNA sequencing of the AVPR2 gene (REFSEQ: NM_000054.6) reveals a heterozygous mutation in exon 2 [p.Val(GTG)88Met(ATG)].
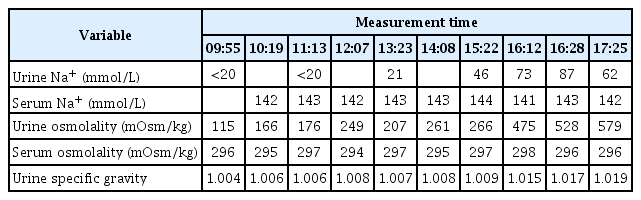
Result of water restriction test
At discharge the patient received desmopressin 0.1 mg (twice a day) as oral medication to be taken during the daytime and underwent follow-up in the outpatient department. Immediately after discharge from the hospital she was transferred to a hospital in her hometown. The effect of desmopressin was not satisfactory, therefore, hydrochlorothiazide was added 2 weeks after discharge.
The patient is currently 4 years and 6 months old; her height is 108.1 cm (75th–90th percentile) and her weight is 20.7 kg (90th–97th percentile); no abnormal developmental problems have been detected. Her intake is approximately 1.5 L with urination 5 to 6 times per day. Although her daily life has not been disrupted, including her attendance in kindergarten, she often urinates in diapers while sleeping without having taken desmopressin.
Discussion
Most cases of congenital NDI, a rare, inherited disorder, are caused by an X-linked recessive AVPR2 genetic mutation. The AQP2 mutation is detected in approximately 10% of patients with NDI who show an autosomal recessive or dominant pattern [3]. The most severe effects are observed in male XL-NDI patients with a hemizygous pathogenic variant in the AVPR2 gene [2].
Many cases of XL-NDI in male patients have been reported, however, symptomatic female carriers with XL-NDI have received far less attention. The patient described here was a symptomatic carrier of XL-NDI who showed a partial response to treatment with desmopressin, suggesting underlying “skewed X-inactivation.” Rapid elevation of the patient’s urine osmolality was observed after administration of desmopressin (0.2 mg, per oral). Genetic testing of AVP for a possible abnormality in AVP itself showed a normal result. Methylation analysis, to confirm the inactivation of the X-chromosome, was skipped as the previous reports show that skewed X-inactivation is always detected in female AVPR2 heterozygotes with XL-NDI [2,4,5].
Some patients with NDI caused by the same gene mutation have been reported to show a partial response to treatment with desmopressin. However, variation in the phenotype of symptoms as well as the patient’s response to desmopressin has been reported [2,3,6,7]. A relationship between this diversity and the degree of X-inactivation has been suggested. The recurrence of fever from infancy was atypical, distinguishing this case from other case reports.
Since the beginning of 2022, the patient's mother has argued that desmopressin has no demonstrable therapeutic effect on her daughter’s daily activities, thereby refusing to allow her to take it. She is currently taking hydrochlorothiazide 25 mg/day in two divided doses. According to her mother, although she is taking the medication, its effect on her symptoms appears to be insignificant. Her mother also said that even though she sometimes forgets to administer the medication, she has not noticed any worsening of her daughter’s symptoms. Discontinuation of hydrochlorothiazide is also under consideration.
The patient described in this case had symptoms that included polyuria, polydipsia, and frequent low-grade fevers. Despite its rarity, complications of congenital NDI can include seizure and retardation of growth and development; therefore, early diagnosis and treatment are critical [7,8]. Pediatricians should suspect NDI in infants who have been diagnosed with polydipsia with frequent episodes of fever.
A typical plan for treatment of patients with NDI includes a low-salt diet to reduce the osmotic load and administration of a thiazide diuretic to enhance reabsorption of salt and water in the proximal tubule. A case involving a patient with the same genetic mutation has been reported, and satisfactory results have been obtained by treatment with thiazide [5]. Pharmacological chaperone therapy studies based on the idea that the expression level and hormone binding affinity of V88M mutation were related to their phenotypic diversity were a failure [6].
Early diagnosis is critical in order to minimize complications. After ensuring an accurate diagnosis, treatment can be administered according to the individual needs of the patient. The current case illustrates the importance of a thorough clinical evaluation along with genotyping for patients who exhibit symptoms prior to making a conclusive diagnosis [7,9].
Few cases involving female patients with partial NDI have been reported. Symptomatic female patients whose diagnoses were based on genetic testing have been reported, however, there are limited data regarding treatment and prognosis [6]. Conduct of studies involving collection of a large number of cases, which include new treatment trials, and collection of data regarding patients’ response to existing treatments should be ongoing.
Notes
Ethical statements
This study protocol was approved by the Institutional Review Board (IRB) of Ajou University Hospital (No. AJOUIRB-EX-2022-396). We were given an exemption from getting informed consent from the IRB.
Conflicts of interest
Hae Il Cheong is an editorial board member of the journal but was not involved in the peer reviewer selection, evaluation, or decision process of this article. No other potential conflict of interest relevant to this article was reported.
Funding
None.
Author contributions
Conceptualization: HIC, KSP
Data curation: JL, KSP
Formal analysis: JL, KSP
Resources: JWL
Supervision: JWL, KSP
Visualization: JL, HIC
Writing-original draft: JL
Writing-review & editing: JL, HIC, JWL, KSP.
All authors read and approved the final manuscript.