Introduction
Alport syndrome (AS) is a progressive inherited kidney disease often accompanied by sensorineural hearing loss (SNHL) and ocular abnormalities such as lenticonus or retinal flecks [1]. Kidney symptoms of AS start with isolated microscopic hematuria, followed by the appearance of microalbuminuria (urine microalbumin-to-creatinine ratio, 30ŌĆō300 mg/g), which progresses to overt proteinuria and a decline in kidney function [2]. AS, which affects one in approximately 5,000 people, is the second most common cause of inherited kidney failure after autosomal dominant polycystic kidney disease [3]. It accounts for 0.5% of new-onset chronic kidney disease (CKD) stage G5 (glomerular filtration rate [GFR] <15 mL/min/1.73 m2 or treatment by dialysis [4]) cases in adults and 12.9% in children [5]. AS is caused by pathogenic variants in the COL4A3, COL4A4, and COL4A5 genes that encode type IV collagen ╬▒3, ╬▒4, and ╬▒5 chains, respectively. Type IV collagen has six different ╬▒ chains, ╬▒1 to ╬▒6, which construct triple-helix structures, the major components of the basement membrane. In the embryonic glomerulus, classical chains of the ╬▒1-╬▒1-╬▒2 triple helix form the glomerular basement membrane (GBM), and with development, these are gradually replaced by novel chains ╬▒3-╬▒4-╬▒5 [6]. Basement membranes of the cochlea and ocular lens have similar type IV collagen compositions as the GBM. Since the novel chains of type IV collagen are defective in AS, basement membranes of the glomerulus, cochlea, and ocular lens become progressively defective [5]. Inheritance patterns of AS are X-linked (XLAS; a defect of COL4A5 on chromosome X), autosomal recessive (ARAS), and autosomal dominant (ADAS; defect of COL4A3 or COL4A4 in chromosome 2), comprising 80%, 15%, and 5%, respectively [1,5].
Inheritance of AS
X-linked AS
Approximately 85% of XLAS patients have a family history of hematuria (with or without proteinuria), kidney failure, or extrarenal manifestations (SNHL or ocular abnormalities). All male patients present with hematuria and early-onset proteinuria, eventually progressing to kidney failure. Ninety percent of patients reached CKD stage G5 by the age of 40 years (median, 25 years) [7]. In males, truncating mutations (rearrangement, nonsense, and frameshift) show a severe phenotype of earlier kidney failure and hearing loss (HL), and non-truncating mutations (in-frame, missense) exhibit a milder phenotype with later kidney failure and HL [7-9]. Hematuria is present in most (>95%) female patients, and proteinuria appears at the median age of 7 years in 75% of females. One-fourth of female patients progress to CKD stage G5 during their lifetime (median age, 65 years), with this progression occurring by the age of 40 years in 15% of them. The phenotype is highly variable in females, ranging from isolated microscopic hematuria with normal kidney function throughout life to kidney failure at a young age [10,11]. SNHL occurs in 90% of males and 12% of females by the age of 40 years [7,10].
Autosomal recessive AS
There are no gender differences in the clinical symptoms, incidence, and prognosis of ARAS. Extrarenal symptoms are common. Both males and females exhibit poor prognoses similar to those with XLAS male patients (the median age of onset is 2.5 years for hematuria, 21 years for developing CKD stage G5, and 13ŌĆō20 years for SNHL) [12,13].
Autosomal dominant AS
In general, both males and females have a good prognosis, and the median ages for developing proteinuria and kidney failure are 17 and 70 years, respectively [5,14]. However, the phenotype is very diverse even within a family, from isolated microscopic hematuria to CKD stage G5. Extrarenal symptoms are uncommon. Their pathologic findings can be either thin basement membrane nephropathy (TBMN) or focal segmental glomerulosclerosis (FSGS). Not rarely, patients with ADAS are misdiagnosed as familial FSGS or IgA nephropathy (IgAN) [14].
Clinical suspicion of AS
The most common symptom of AS is hematuria, which is persistent in 100% of XLAS male and ARAS patients, and may appear intermittently in approximately 95% of XLAS females and 50% of ADAS patients (GeneReviews). Recurrent gross hematuria (especially following upper respiratory infection) occurs in infancy or early childhood in 40% to 60% of cases, with an average age of 3.5 years in males and 9 years in females. Males without hematuria by the age of 10 years are unlikely to have AS [15]. Importantly, proteinuria does not appear without hematuria [16]. Ocular abnormalities of AS are less sensitive than the associated HL; however, they are more specific, which means that they could be diagnostic. Lenticonus and central fleck retinopathy occur only in AS and are associated with kidney failure before 30 years in male patients with XLAS [17].
Histologic findings
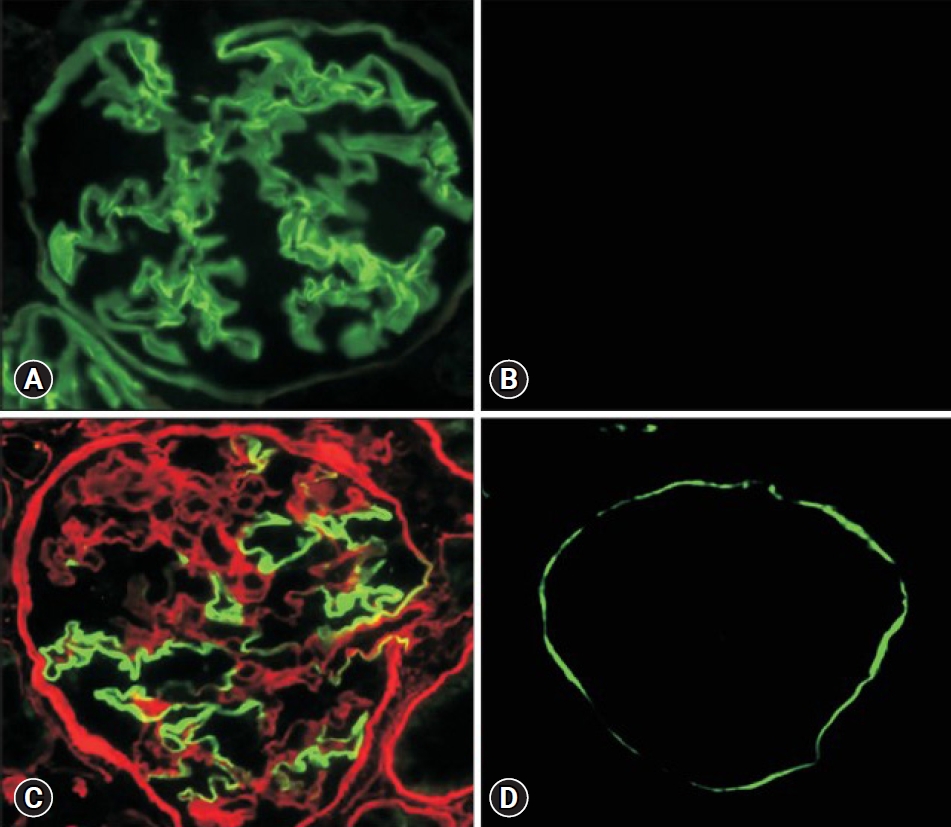
Conventionally, the diagnosis of AS was made pathologically; however, this can be challenging. There are no specific light microscopic findings in AS; mesangial proliferation, FSGS, and interstitial infiltration containing lipid-laden foam cells may be observed [18,19]. Electron microscopy findings typically show irregular thickening and thinning of the GBM, lamellation, and splitting in the lamina densa of a "basket-weave" appearance (Fig. 1) [20]. While these findings are very characteristic of AS, they appear later in the course of the condition; therefore, electron microscopy may show only diffuse GBM thinning in young male patients with XLAS or XLAS females, or ARAS/ADAS patients [5,21]. Immunofluorescence staining of type IV collagen ╬▒5 can be diagnostic, irrespective of the patientŌĆÖs age. Typically, collagen type IV ╬▒5 chain expression is completely absent in XLAS male patients (both at GBM and Bowman capsule) and ARAS (GBM) patients; however, more than 20% of XLAS males and 20% of ARAS patients exhibit normal expressions of ╬▒5 if their pathologic variants are non-truncating [13,22], as well as ADAS patients. XLAS female patients exhibit a mosaic pattern of ╬▒5 staining (Fig. 2) [1]. The collagen type IV ╬▒5 stain in skin biopsy is valid only for XLAS since the epidermal basement membrane is mainly comprised of the triple helix of ╬▒5-╬▒5-╬▒6 as the Bowman capsule [23]. Therefore, the normal expression of collagen type IV ╬▒5 AS cannot exclude AS. Furthermore, nonspecific focal GBM thinning may also be seen in IgAN [24]. Thus, kidney biopsy plays a supportive, not confirmatory role in the diagnosis of AS [25].
Advances in the last decade
Due to these limitations of histopathology and the remarkable development of molecular-based techniques in the past decade, genetic testing has become the first-line diagnostic technique for AS. Because COL4A genes are large, targeted next-generation sequencing, including all three novel chain genes, has become the primary genetic screening method instead of Sanger sequencing [3,26]. Although there is currently no curative therapy for AS, nephroprotective drugs such as angiotensin-converting enzyme inhibitors (ACEi) and angiotensin receptor blockers can delay the progression to kidney failure for years or even decades. In a large cohort study of XLAS males in Japan [27], kidney replacement therapy (KRT) was delayed by 17 years and 12 years in the non-truncating and truncating mutant groups, respectively, in the group of patients treated with either ACEi or angiotensin receptor blockers compared to those in the untreated group. In addition, multiple prospective and retrospective studies have reported the beneficial effects of early interventions with ACEi. The 2020 guideline emphasizes early molecular diagnosis to enable early treatment and avoid immunosuppression [2].
The rationale for early ACEi treatment
In a study using the mouse model of ARAS, those treated with ramipril at 4 weeks survived twice as long as mice not treated with ramipril, whereas the group that started treatment at 7 weeks did not differ in survival compared to the untreated group [28]. A retrospective cohort study in Europe showed that among those who began treatment when they had only isolated hematuria or microalbuminuria, no one needed KRT until the age of at least 40 years [29]. Treatment initiation when patients had proteinuria (>0.3 g/day) with normal kidney function delayed KRT for 18 years (median age of KRT, 40 years). Later initiation at CKD stage G3 (GFR, 30ŌĆō59 mL/min/1.73 m2) or G4 (GFR, 15ŌĆō29 mL/min/1.73 m2) had the effect of postponing KRT by only 3 years (median age, 25 years) compared to the untreated group with a median KRT age of 22 years. Based on these studies, the Early Prospective Therapy European Community Trial in Alport syndrome (EARLY PRO-TECT Alport) was conducted in Germany to determine whether starting ramipril treatment on noticing isolated hematuria or microalbuminuria could delay the progression of the disease to its next phase (microalbuminuria or proteinuria, respectively). Early treated group (n=11, open-label [n=42]) developed disease progression in 27%, 41%, for each, whereas the placebo group (n=9) in 56%. Although not statistically significant, the authors concluded that the early initiation of ramipril might be beneficial in delaying progression by more than 40% [30].
Expanded phenotypes of AS
With the broad implementation of next-generation sequencing, several cases are rediagnosed with AS even when their symptoms are not always compatible with AS. In a large-scale analysis of whole-exome sequencing in 3,315 patients with CKD of all causes, 10% were found to have genetic causes, of which 30% were diagnosed as AS with COL4A mutations. The majority of these patients (62%) had been misdiagnosed with hypertensive nephropathy, other kinds of glomerulonephropathy such as steroid-resistant nephrotic syndrome, IgAN, or FSGS [20,31]. COL4A defects were the most common causes in adult-onset FSGS, accounting for up to 20% of familial FSGS [32]. Therefore, the COL4A3-COL4A5 mutations must be considered in CKD of unknown etiology, steroid-resistant nephrotic syndrome, FSGS, and IgAN, even if the clinical manifestations and biopsy findings are not fully compatible with AS, especially if there is a contributive family history [26]. Recent guidelines emphasize that FSGS or IgAN with pathogenic variants of COL4A should be considered ŌĆ£ASŌĆØ and not as ŌĆ£coincidental diseaseŌĆØ [2].
The new classification scheme for AS
From ŌĆ£carrierŌĆØ to ŌĆ£patientŌĆØ in XLAS females
Traditionally, females with COL4A5 heterozygous mutations have been considered ŌĆ£carriers.ŌĆØ While previously regarded as benign, 25% of them develop CKD stage G5 during their lifetime. Risk factors for progression include a history of gross hematuria in childhood, proteinuria, nephrotic syndrome, SNHL, and the presence of diffuse thickening and lamellation of the GBM. Therefore, experts stressed that females with COL4A5 heterozygous mutations should be considered ŌĆ£patientsŌĆØ rather than benign ŌĆ£carriers.ŌĆØ Female AS patients need to be monitored regularly for proteinuria and kidney function [33,34].
From ŌĆ£TBMNŌĆØ and ŌĆ£ADASŌĆØ
TBMN, traditionally known as ŌĆ£benign familial hematuria,ŌĆØ is characterized by hematuria with or without mild proteinuria, with diffuse GBM thinning. This condition was considered a benign one that does not progress to CKD stage G5. However, TBMN, in many cases [35], is caused by heterozygous mutation of COL4A3 or COL4A4, same as ŌĆ£ADAS,ŌĆØ and there had been no definite distinction [5]. Moreover, the progression to CKD stage G5 in TBMN has been reported, especially in patients with risk factors of proteinuria, FSGS, or GBM thickening and lamellation on kidney biopsy, SNHL, and family history of progressive kidney disease [33,36]. So, is TBMN equal to ADAS? A guideline published in 2019 did not agree, as the likelihood of kidney failure is minimal [3]. However, the latest guidelines emphasize that all diseases caused by heterozygous mutations of COL4A3 or COL4A4 should be classified as ŌĆ£ADASŌĆØ for an earlier and more aggressive intervention since earlier ACEi treatment delays kidney failure for longer [2,33]. In a recent study, Yamamura et al. [37] reported that ADAS accounted for 17% of all AS cases, which is much higher than the 5% found in previous reports, suggesting that the diagnosis of ADAS is underestimated [37]. Therefore, even when diagnosed with TBMN, especially if associated with COL4A3 or COL4A4 variants, regular follow-up is strongly recommended for the risk of kidney failure.
Early diagnostic tactics
Early genetic test necessity
The latest guideline proposed in 2020 by Kashtan and Gross [2] recommends that genetic testing should be performed if AS is suspected in patients with persistent glomerular hematuria. Kidney biopsy is recommended if genetic testing has uncertain pathogenicity and AS is not suspected based on clinical data or family history. Kidney biopsy should include transmission electron microscopy, and if transmission electron microscopy is not available, immunofluorescence of the type-IV collagen stain is required.
Treatment guidelines
When should we start treatment and how do we monitor it?
The clinical practice recommendations from the Alport Syndrome Research Collaborative emphasize initiating early interventions. In XLAS male and ARAS patients, who progress to CKD stage G5 in 100% of cases, treatment should be started as soon as possible when the diagnosis is obtained in patients older than 1 year regardless of proteinuria (strong recommendation). For XLAS female and ADAS patients, treatment initiation is recommended if microalbuminuria develops during annual monitoring [2]. This differs significantly from their previous guideline in 2013 that did not recommend treatment with isolated hematuria and recommended optional treatment with microalbuminuria in XLAS males [38].
Which drugs should we use?
The drug recommended by the above-mentioned guideline is either ramipril or lisinopril. Ramipril is a well-established drug with evidence of efficacy and safety, which was adopted from protocols in the ESCAPE (Effect of Strict blood pressure Control and ACE Inhibition on the Progression of CKD in Pediatric Patients) trials [39] and the EARLY PRO-TECT trial for children with AS [30]. Lisinopril is another ACEi with a comparable duration of action to that of ramipril, and it has obtained evidence from some pediatric studies [40,41]. In the ARAS mouse model study, ramipril prolonged the lifespan significantly more than candesartan did (111% vs. 38%, respectively) [42]. However, no AS studies in humans have compared ramipril with other drugs. One small study compared the antiproteinuric effects of enalapril to those of losartan in children with AS and found no significant difference between the two [43].
How to increase the dose of the ACEi?
It is emphasized that ACEi should be up titrated more rapidly and aggressively than previously recommended. The previous guideline in 2013 recommended increasing ramipril dose ŌĆ£everyŌĆØ 3 months until the target urine protein-to-creatinine ratio (UPCR; less than half of the baseline value) is reached [38]. However, the recent guideline in 2020 recommends increasing the dose ŌĆ£overŌĆØ the first 3 to 4 months starting 1 mg/m2/day to a maximum of 6 mg/m2/day of ramipril irrespective of the degree of proteinuria if the patient can tolerate such an increase. The dosage needs to be increased as the child grows to maintain the maximum dose [2].
Dual renin-angiotensin-aldosterone system blockade
Data from the ESCAPE trial in pediatric CKD reported that a UPCR <1.0 mg/mg was associated with better kidney outcomes [44]. Therefore, the latest guideline suggested that considering the use of dual renin-angiotensin-aldosterone system (RAAS) blockade may be reasonable if the UPCR exceeds 1.0 mg/mg despite the maximum tolerated dose of ramipril or lisinopril. Losartan can be added in small amounts, at an initial dose of 0.8 mg/kg/day [2]. However, there is limited evidence of the efficacy and safety of dual RAAS blockade, and the Kidney Disease: Improving Global Outcomes (KDIGO) guideline recommends not using dual blockage in another glomerulopathy. When using dual RAAS blockade, adverse effects such as hyperkalemia, kidney insufficiency, and hypotension should be periodically monitored [2,45].
Cyclosporine
Cyclosporine (CsA) is not recommended in patients with AS [2]. CsA diminishes proteinuria by directly stabilizing the podocyte cytoskeleton [46]; however, the long-term use of this drug may stimulate profibrotic mediators such as transforming growth factor-beta, leading to interstitial fibrosis and tubular atrophy [47]. In small, uncontrolled studies conducted in Spain [48,49], France [50], and Italy [51], proteinuria significantly decreased throughout CsA treatment (5 mg/kg/day). However, the effect was temporary: proteinuria nearly returned to the baseline value after discontinuation in most patients, and kidney outcomes were conflicting with stable GFRs over 8 years [48,49] versus kidney function decline over 6 months, and some patients developed interstitial fibrosis 20 to 27 months after CsA initiation [50].
Hearing and ophthalmologic evaluation and follow-up
HL in children, even in mild cases, affects speech-language, social-behavior, cognitive development, and academic performances [52]. Several papers have shown that HL in AS is sensorineural, progressive, and bilateral, often affecting the middle and particularly high frequencies [53]. Therefore, it can only be detected by formal hearing tests, especially during early childhood. Approximately 30% of XLAS males and 20% of ARAS patients exhibit detectable HL by age 10. In males with XLAS, HL increases to approximately 60% by age 20 and shows a genotype-phenotype correlation. ARAS patients show a high rate of HL when they have one or more truncating mutations [8,13,54]. Therefore, a hearing evaluation is recommended annually for XLAS males and ARAS, starting at the age of 5 to 6 years, and earlier if overt proteinuria occurs or symptoms suggestive of HL, such as speech delay, develop. For XLAS females, the probability of HL is less than 10% by the age of 40 years; however, HL eventually occurs in 30% of them [10]. It is recommended that females with XLAS perform a formal hearing test when overt proteinuria is present. All AS patients should avoid loud sounds. If HL develops, it usually responds well to hearing aids [2].
Ocular abnormalities in AS, lenticonus, and central fleck retinopathy have high diagnostic and prognostic values [17]. For XLAS males with truncating mutations and ARAS patients, it is strongly recommended that ocular investigations begin at age 15 (earlier if they have an abnormal vision) and annual check-ups should be performed. For females with XLAS and ADAS patients, ophthalmologic assessments are recommended if clinically indicated [2].
Other recommendations
Hypertension can accelerate the deterioration of kidney function. Therefore, it should be strictly controlled, and the target blood pressure should be in the 50th percentile [25]. As lifestyle modifications, maintaining a body mass index of <25 kg/m2, moderating oneŌĆÖs dietary intake of meat protein and salt, and avoiding smoking are also recommended [2].
Research horizons
Future therapeutic options
In a clinical trial on AS-induced CKD [55], bardoxolone, an activator of nuclear factor erythroid-related factor 2 (Nrf2), improved the GFR. However, another study of this drug in patients with type 2 diabetes mellitus and CKD stage G4 (BEACON trial) was prematurely terminated due to fatal cardiovascular complications (relative risk, 1.83; P<0.001) [56]. For safety and efficacy reasons, the Food and Drug Administration recently denied the approval of the drug in AS.
Conclusions
Advances in molecular genetics and clinical studies over the past decade have made early diagnosis and intervention possible in patients with AS. Also, histopathologic diagnoses other than AS cannot exclude AS, since some of such patients harbor disease-causing variants of AS. Therefore, even when the clinical phenotype and biopsy results are not compatible with AS, the COL4A3-COL4A5 mutations need to be considered, especially if there is a contributive family history. Female subjects affected by XLAS are no longer considered ŌĆ£carriersŌĆØ but ŌĆ£patients,ŌĆØ and individuals with heterozygous mutations in COL4A3 and COL4A4 (even diagnosed with ŌĆ£TBMNŌĆØ) should be monitored regularly due to the risk of kidney failure. XLAS males and ARAS patients older than 12 months need to undergo ACEi regardless of proteinuria, while other types can be monitored for microalbuminuria appearance, which indicates treatment.

 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print