Introduction
Hemolytic uremic syndrome (HUS) is a form of thrombotic microangiopathy (TMA), characterized by microangiopathic hemolytic anemia, thrombocytopenia, and acute kidney injury [1]. In children, enterohemorrhagic Escherichia coli (EHEC) producing Shiga toxin is the most common cause of HUS (typical HUS), accounting for 90% of pediatric cases. However, some cases are associated with inherent dysregulation of the complement system (atypical HUS, aHUS), commonly caused by mutations in components of the complement system, including factor H (CFH), factor I, factor B, or complement 3 [2]. In the past, plasma therapy (plasma exchange or plasma infusion) was applied to ameliorate the dysregulation of complement activation, which is often insufficient to prevent permanent damage to the kidneys. Currently, monoclonal humanized anti-C5 antibodies, such as eculizumab, which block activation of the complement pathway, are the first-line treatment for aHUS with excellent renal outcomes [3]. However, C5 blockage is not always safe because the complement system plays a crucial role in the immune system, as indicated by the fatal outcome of meningococcal infection in patients who were treated with eculizumab, the first monoclonal humanized anti-C5 antibody approved for the treatment of aHUS [4]. Therefore, C5 blockade is only indicated when dysregulated complement activation is involved in the pathophysiology of aHUS.
While CFH mutations are the most common cause of aHUS, especially in children [5,6], genes other than those involved in the complement system have also been implicated in aHUS. DGKE, encoding diacylglycerol (DAG) kinase epsilon (DGKE), is one such gene. Lemaire et al. [7] identified this gene using whole-exome sequencing of a patient with infantile aHUS. DGKE is found in the endothelium, platelets, and podocytes. In endothelial cells, arachidonic acid-containing DAG activate protein kinase C, promoting thrombosis, and DGKE normally inactivates DAG signaling [7]. Therefore, DGKE mutations result in a thrombogenic status, which is not related to complement pathway activation. DGKE mutations are known to cause steroid-resistant nephrotic syndrome or membranoproliferative glomerulonephritis [8]. In Republic of Korea, an aHUS case caused by a DGKE mutation has been reported previously [6], but the details of the clinical course are not well documented.
Here, we report the second case of aHUS caused by DGKE mutations in Republic of Korea.
Case report
An 11-month-old male patient presented to a local pediatric clinic with a fever. Antibiotics were prescribed for the presumptive diagnosis of acute pharyngitis. A few days later, he developed diarrhea followed by vomiting and hematuria and was transferred to our institution. At our hospital, he had hypertension with a systolic blood pressure above 140 mmHg; he looked acutely ill and anemic. His tongue and lips were dehydrated, and he was edematous, especially in his extremities and eyelids. Laboratory workup showed anemia (hemoglobin [Hb], 6.3 mg/dL), thrombocytopenia (79,000/╬╝L), high blood urea nitrogen (94 mg/dL), high creatinine (1.99 mg/dL), high lactate dehydrogenase (3,297 IU/L), hyperuricemia (14.2 mg/dL), hyperphosphatemia (7.2 mg/dL), high urine protein/creatinine ratio (UPCR; 16.14 g/g creatinine), and hematuria (50ŌĆō99 red blood cells per high-power field [RBC/HPF]). His complement 3 level (112 mg/dL) and 4 level (18 mg/dL) were within normal limits. EHEC test result was negative and there was no stool Hb. On kidney ultrasonography, the parenchymal echogenicity of both kidneys increased without hydronephrosis. He had oliguria (80 mL/day, 7.8 mL/kg/day); thus, hemodialysis was administered for 1 day. Amlodipine, allopurinol, and calcium carbonate were prescribed for hypertension, hyperuricemia, and hyperphosphatemia, respectively. There was no laboratory evidence of EHEC infection, but the presumptive diagnosis was typical HUS because he had prodromal diarrhea. After seven days, almost all laboratory results improved with conservative management, similar to typical HUS. However, he was very young and did not have a history of raw food intake that could have caused EHEC infection. Therefore, the patient was discharged with a warning of recurrence and suspected aHUS. When he was 26-month-old, 15 months after the first episode, he had a fever of up to 39┬░C and melena. The fever subsided after 2 days, but he looked pale after 4 days, so he visited a local pediatric clinic. Hematuria, proteinuria, and anemia (Hb, 9.8 mg/dL) were found; therefore, he was transferred to our institution. In laboratory workup, mild anemia (Hb, 9.4 mg/dL) and mild creatinine elevation (0.42 mg/dL, baseline 0.35 mg/dL) were noted along with elevated plasma Hb (14.6 mg/dL) and lactate dehydrogenase (585 IU/L), suggesting hemolysis. Hematuria (>100 RBC/HPF) and proteinuria (UPCR, 5.60 g/g creatinine) were evaluated by urine analysis. Both stool polymerase chain reaction and culture were negative for EHEC. After admission, the patientŌĆÖs general condition and laboratory abnormalities improved without treatment for several days. Suspecting an aHUS relapse, a kidney biopsy was performed. The glomeruli were mildly increased in size and had focal mild hypercellular endothelial cells and tram-track appearance. Two global sclerotic glomeruli were noted among the 57 glomeruli. Slight focal infiltration of mononuclear cells was observed in the tubules. Diffuse thickened glomerular basement membrane, slight focal effacement of the foot process, subendothelial widening and mesangial interposition were observed by electron microscopy. In immunofluorescence staining, C3 and Lambda were reported as +/ŌĆō, and IgM and C4d were reported as positive in the glomerular capillary loops and peritubular capillaries (Fig. 1). Therefore, the pathological findings were consistent with chronic TMA. The TMA gene panel revealed a homozygous nonsense mutation (c.1498C>T in exon11 (p.Arg500*)) in DGKE. This gene panel covers 25 genes associated with TTP and HUS (ADAMTS13, C1S, C2, C3, C5, C8A, C9, CD55, CD59, CFB, CFD, CFH, CFHR5, CFI, CR2, DGKE, F12, INF2, MASP1, MASP2, MMACHC, MMUT, PLG, THBD, WT1).
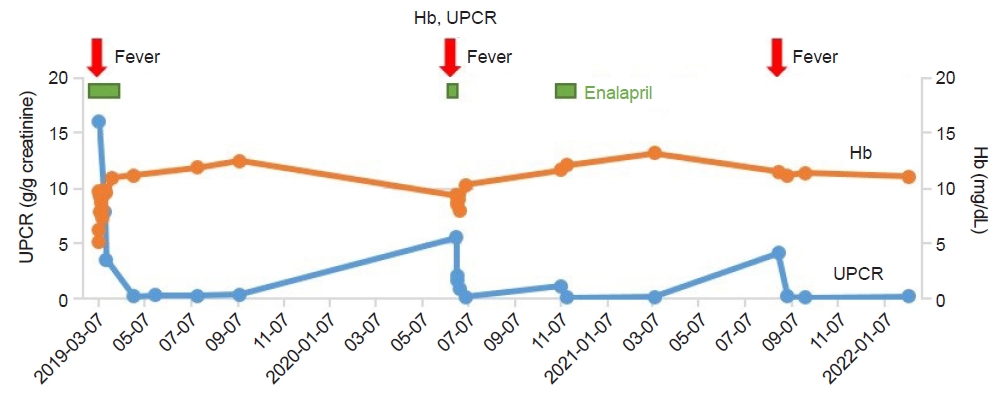
Proteinuria was monitored during follow-up. When he was 31-month-old, his proteinuria increased to 1.17; therefore, enalapril was prescribed. After 2 weeks, proteinuria disappeared and he did not recur despite discontinuation of medications. At the age of 40 months, 29 months after the first episode, he experienced a third episode of aHUS along with a fever of up to 40┬░C and hematuria. Spontaneous remission was achieved within 1 month without medication. At the last follow-up at the age of 46 months, his blood pressure and laboratory findings were unremarkable, without proteinuria. Hb and UPCR levels during follow-up are displayed in Fig. 2.
Discussion
This is a case of recurrent HUS that showed spontaneous remission with supportive care. Because of the infantile-onset and relapse history, aHUS was suspected, and a DGKE mutation was identified by a genetic test. The homozygous nonsense mutation of this patient (c.1498C>T in exon11 (p.Arg500*)) has not been reported before. However, as it is a truncating mutation, the mutation is considered as pathogenic in this patient. Similar to previous reports on DGKE mutations, our case presented at a very early onset (median age <1 year) with aHUS with a self-limiting disease course [9,10]. His initial presentation was accompanied by diarrhea; therefore, typical HUS was suspected at first. However, aHUS is often triggered by infection, and the first episode in our case was triggered by gastrointestinal infection.
In general, HUS in young children is typically followed by bloody diarrhea due to EHEC infection. Typically, they have a history of ingesting raw or undercooked food. Otherwise, aHUS should be suspected, especially in very young infants. Unlike typical HUS, aHUS does not spontaneously remit and often relapses. Since it can be fatal, aggressive management is necessary, previously with plasma and now with C5 inhibitors, if aHUS is associated with complement dysregulation. The CFH mutation was first suspected in a case of very young aHUS. For aHUS with a CFH mutation, C3 levels often decrease, and approximately 60% to 70% of patients lose renal function if not properly managed [11]. However, our patient had normal C3 levels and a self-remitting course, which was not consistent with aHUS associated with complement dysregulation. In such cases, a DGKE defect must be suspected. Currently, correct genetic diagnosis is more important because of the availability of C5 inhibitors, the treatment of choice for complement-related aHUS.
Eculizumab, the currently available C5 inhibitor, is an antibody targeting the complement pathway; it is unrelated to the DGKE mutation, which is related to the coagulation pathway. There have been some case reports of DGKE mutation-associated aHUS in which eculizumab was effective [12]. However, these cases might have recovered even without eculizumab, since DGKE-associated aHUS is usually self-remitting. Eculizumab has been proven to be relatively safe and very effective for aHUS, but it is regularly administered to prevent relapse of aHUS once indicated. Therefore, even when aHUS is suspected, causes other than complement system dysregulation must be considered before deciding to administer C5 inhibitors. Other than DGKE mutations, secondary causes of aHUS include medication, malignancy, infection, autoimmune diseases, and genetic causes, such as cobalamin C defect or G6PD deficiency.
Despite the self-remitting course of aHUS caused by DGKE defects, the long-term outcome of DGKE defects is not favorable. Chronic kidney disease stages 4 and 5 are common in patients with DGKE mutation [7]. Until the last follow-up, our patient showed third relapse. Chronic relapse of aHUS or development of membranoproliferative glomerulonephritis and/or steroid-resistant nephrotic syndrome might occur in this patient in the future. Therefore, careful long-term follow-up was indicated in this case.
HUS in infants is not common, mandating the suspicion of aHUS. While CFH or other complement-related aHUS is more prevalent, DGKE mutations need to be suspected in cases with normal complement levels and spontaneously recovering courses. Although aHUS related to DGKE may recur, a C5 inhibitor is not indicated. However, close follow-up is necessary, because other glomerulopathies may have occurred in this case.

 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print