Severe Hypophosphatemia in a Girl with Systemic Lupus Erythematosus
Article information
Abstract
Systemic lupus erythematosus (SLE) is characterized by various symptoms and multi-organ involvement. Hypophosphatemia has been described in several diseases accompanied with systemic inflammation. However, hypophosphatemia has rarely been described in SLE patients, especially in those without nephritis. We report the case of a 13-year-old girl with SLE who developed hypophosphatemia without renal involvement. Her hypophosphatemia was caused by renal loss of phosphorus and persisted for 7 months. It improved as her complement levels increased. Therefore, hypophosphatemia may be related to disease activity in SLE patients.
Introduction
Systemic lupus erythematosus (SLE) is a systemic autoimmune disorder characterized by various symptoms, such as fatigue, fever, myalgia, and arthritis, with multi organ involvement. Among the various organs, renal involvement is significant and can lead to morbidity and mortality [1].
Hypophosphatemia has been reported in several autoimmune disorders, such as Sjögren’s syndrome, tumors, and thyroiditis, and also in sepsis [2-5]. However, it has rarely been described in SLE.
Here, we report a case of hypophosphatemia in a SLE patient without nephritis, which was caused by the renal loss of phosphorus, it improved as disease activity improved.
Case report
A 13-year-old girl with myalgia for 1 month was referred to our hospital. A week ago, she was admitted to a local hospital for lethargy and myalgia. One week before admission, she experienced eczema. During admission, mild fever was checked twice, and leukopenia, abnormal liver function, and thrombocytopenia were found.
On physical examination, her blood pressure was 99/68 mmHg and her pulse rate was 125 beats/min. She showed myalgia, general weakness, oral ulcers, vasculitic lesions on her face, weight loss, and hair loss. She did not have joint pain, photosensitivity, or the Raynaud’s phenomenon. There were no signs of renal involvement, such as proteinuria, hematuria, and azotemia. The initial laboratory findings are shown in Table 1.

Details in Initial Laboratory Test
She satisfied 5 of the 11 American College of Rheumatology 1997 Revised classification criteria for SLE, discoid rash, oral ulcers, leukopenia, positive anti-double stranded deoxyribonucleic acid antibody (anti-dsDNA) and positive antinuclear antibody (ANA) [6]. Also, a diagnosis of SLE was made by disocoid rash, oral ulcers, hair loss, leukopenia, positive ANA, positive anti-dsDNA antibody, low complement levels and positive direct Coombs' test, fulfilling 8 of the 17 Systemic Lupus International Collaborating Clinics classification criteria for SLE [7]. Her Systemic Lupus Erythematosus Disease Activity Index (SLEDAI) score was 12. The treatment was initiated with prednisolone and hydroxychloroquine. Her symptoms gradually improved after treatment, but the complement levels remained low. In addition, hypophosphatemia without hypercalcemia or abnormal level of intact parathyroid hormone was observed. Therefore, we measured the reabsorption of phosphorus from renal tubule.
The tubular reabsorption of phosphorus (TRP) was 89.7%, which was within the normal range. Also, the ratio of the tubular maximum reabsorption of phosphate to glomerular filtration rate (TmP/GFR) was 1.61 mg/dL, which was extremely low compared with the normal range. A few months after the diagnosis, we started to treat her by oral phosphate replacement and hypophosphatemia was improved after recovering the normal complement level. Seven months after the treatment, we stopped oral phosphate replacement and her level of phosphorus became normal.
Twenty-one months since starting treatment, she has maintained a normal phosphorus range without replacement, and the prednisolone dose is being reduced. The treatment and disease course of our patient are shown in Fig. 1.
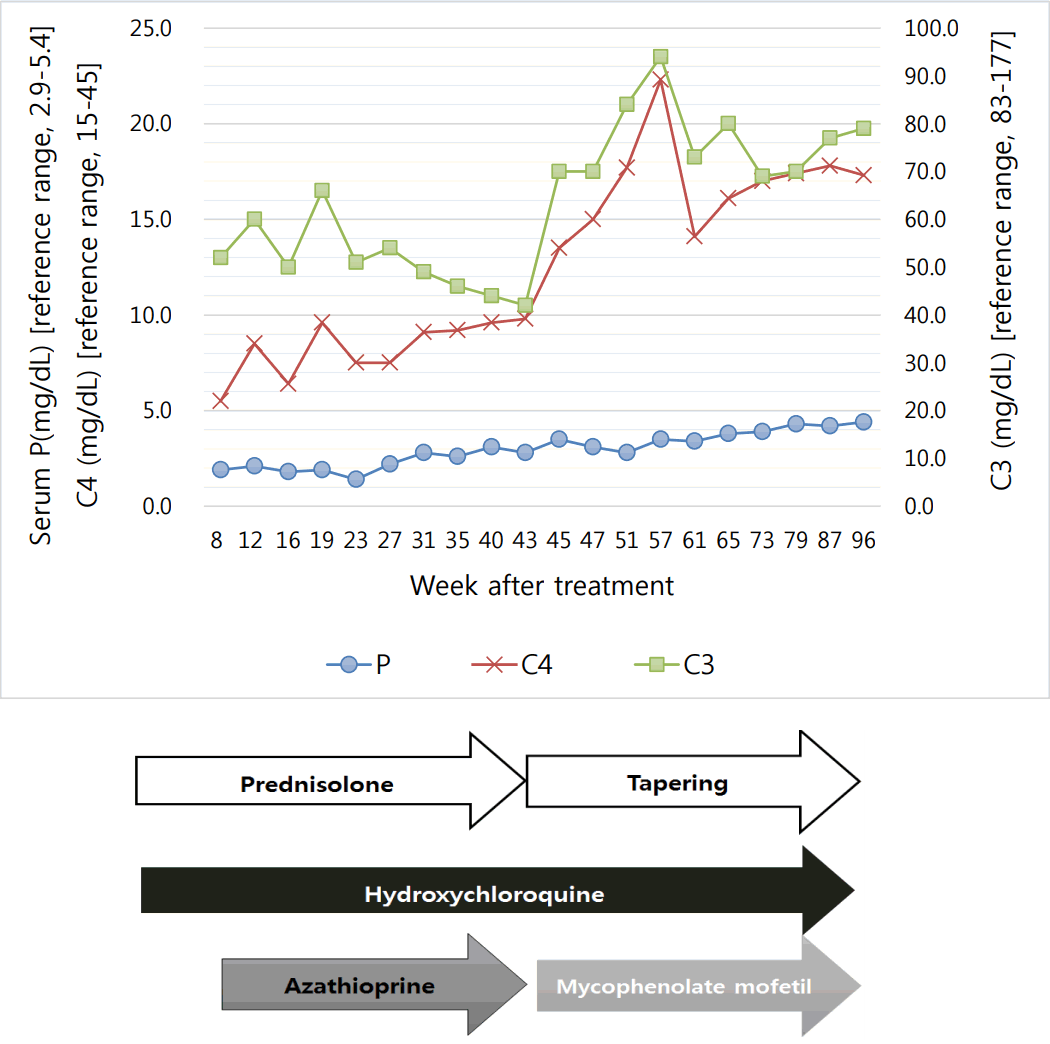
Treatment and disease course of the patient. P, phosphorus; C3, complement 3; C4, complement 4.
Discussion
This case is the first report of hypophosphatemia in SLE patient in Korea. Hypophosphatemia in SLE has rarely reported. Further, there were no other abnormalities, such as hypomagnesemia, hyponatremia, hypercalcemia, or hyperparathyroidism. Fujiwara et al. reported that hypophosphatemia in SLE is associated with high serum tumor necrosis factor α (TNF- α) and interleukin 6 levels (IL-6) [8]. Before this report, there had been several reports about cytokine and hypophosphatemia. Zwaveling et al. reported them in cancer, and Barak et al. reported them in early sepsis [3,5]. Recently, Yamazawa et al. reported them in macrophage activation syndrome [9]. All of them have paid attention to cytokines, such as TNF- α and IL-6, as a cause of hypophosphatemia. TNF- α is known to affect the reabsorption of sodium, phosphates, uric acid, glucose, and bicarbonates at the proximal tubules [10]. Therefore, hyponatremia, hypouricemia, and metabolic acidosis are accompanied with hypophosphatemia. In our case, however, except for hypophosphatemia, there were no other abnormal laboratory findings. Therefore, the patient’s pathogenesis was not clear.
Most reports describe cytokines, but unfortunately, we did not investigate the patient’s cytokines, which made it difficult for us to describe the pathogenesis of hypophosphatemia. However, we found that disease activity is associated with hypophosphatemia. As we showed in Fig. 1, the complement level was correlated with the phosphorus level. It seems that the phosphorus level may suggest disease activity in SLE patients.
Hypophosphatemia caused by the renal loss of phosphorus has been described in inherited disorders, such as X-linked hypophosphatemia [11]. Renal sodium-phosphate cotransporter is an essential part of these disorders [12]. Fibroblast growth factor 23 is one of the representative genes that regulate this transporter [13]. Recently, sodium-hydrogen exchanger regulatory factor 1 was reported to be a regulator gene [14]. However, as the patient’s hypophosphatemia improved, she could not have had gene mutation. Instead, we can presume that there are unknown substances that affect the cotransporter and that may be related to SLE activity
In summary, our case of a 13-year-old girl with SLE exhibited hypophosphatemia, without hypomagnesemia, hyponatremia, hypercalcemia, or hyperparathyroidism. The cause was the loss of phosphorus from the kidney, which was confirmed by TRP and TmP/GFR. Further works involving a large number of patients from a multicenter are needed.
Notes
Conflict of interest
No potential conflict of interest relevant to this article was reported.