Introduction
Primary Epstein-Barr virus (EBV) infections are common in infants and children. In most cases, patients are asymptomatic or have non-specific symptoms [1]. Less commonly, they are associated with severe complications such as hemolytic anemia, pancytopenia, myocarditis, hepatitis, splenic rupture, glomerulonephritis, hemophagocytic syndrome, Guillain-Barr├® syndrome, meningitis, encephalitis, and psychiatric disorders [1]. Henoch-Sch├Čnlein purpura (HSP) can result from exposure to an antigen after infection with several types of organisms. In rare cases, HSP can result from EBV infection [2].
Here, we report the case of a 32-month-old female patient with primary EBV hepatitis manifesting as HSP.
Case report
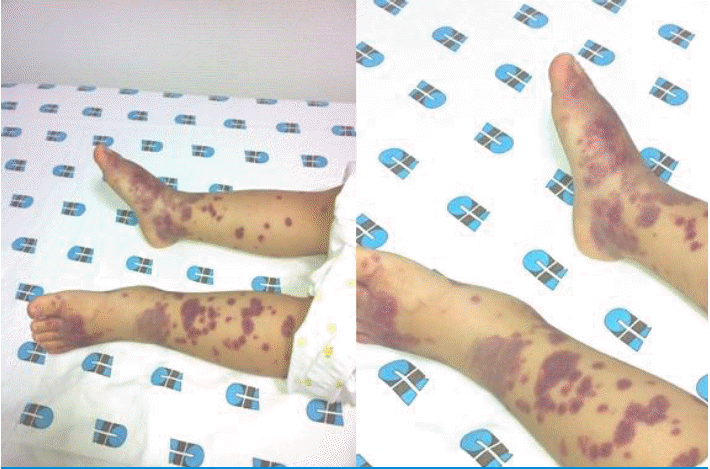
A 32-month-old female was admitted to our hospital because of a 1-day history of fever, arthralgia, and swelling in both ankles for 1 day, and purpura on both legs and arms. She had passed grayish stools intermittently for a few days previously. The patient's medical history was unremarkable. Vital signs included a blood pressure of 80/50 mmHg, heart rate of 110/minute, respiratory rate of 25/minute, and body temperature of 38.5Ōäā, and she did not look ill, although she seemed tired. A physical examination revealed multiple palpable purpuric lesions on the lower limbs and fewer purpuric lesions on the upper limbs. Both feet and ankles were slightly swollen and tender (Fig. 1), and both ankles and knees showed slight limitation of motion. There was no pharyngeal injection, conjunctivitis, or tongue lesion, and no palpable cervical, axillary, or inguinal lymph nodes were detected. The abdomen was soft, flat, and not tender. The liver and spleen were not palpable. Bowel sounds were normoactive. Laboratory findings were as follows: hemoglobin 12.4 g/dL, white blood cells 15,860/mm3 (seg 62%, lymphocytes 25%, monocytes 11%, atypical lymphocytes 2%), platelet count 103,000/mm3, normocytic, normochromic, acanthocytes 1+ on a peripheral blood smear, reticulocytes 1.07%, C-reactive protein 6.5 mg/dL, erythrocyte sedimentation rate 9 mm/h, protein 6.7 g/dL, albumin 4.1 g/dL, glucose 137 mg/dL, blood urea nitrogen/creatinine 15.4/0.6 mg/dL, T-bilirubin 1.23 mg/dL (D-bilirubin 0.64), glutamic-oxaloacetic transaminase (GOT)/glutamic-pyruvic transaminase (GPT) 910/1,244 IU/L, alkaline phosphatase 1,696 IU/L, r-glutamyl transpeptidase 166 U/L, creatine kinase 61 U/L, lactate dehydrogenase 1,753 U/L, ammonia 27 ┬Ąg/dL, cholesterol 22.7 mg/dL, amylase 40 U/L, serum electrolytes Sodium 130 mEq/L, potassium 4.8 mEq/L, chloride 98 mEq/L, TCO2 22.7 mEq/L, Prothrombin time 15.5 s, international normalized ratio 1.4, activated partial thromboplastin time 36.4 s, D-dimer 2,138.04 ng/mL, fibrinogen 326 mg/dL, C3 117 mg/dL, C4 47 mg/dL, fluorescent antinuclear antibody (-), anti-neutrophilic cytoplasmic antibodies (-), IgG 959 mg/dL, IgA 101 mg/dL, IgM 125 mg/dL, Mycoplasma pneumonia IgM negative, cold agglutinin negative, direct/indirect Cooms test negative/negative, and rheumatoid factor negative. The results of urinalysis were normal, as were those of urine and blood cultures and chest radiography (posterioranterior view). Simple examination of the abdomen showed suspicious hepatosplenomegaly (Fig. 2), but abdominal sonography revealed no remarkable findings. Tests for viral markers revealed surface antigen of the hepatitis B virus negativity, but antibody to hepatitis B surface antigen positivity. Polymerase chain reaction confirmed the presence of EBV DNA. EBV viral capsid antigen (VCA) IgM antibodies but not IgG antibodies were detected in serum. Tests for other viral markers yielded negative results (Hepatitis A virus, Hepatitis C virus, Hepatitis E virus, Herpes simplex virus, Cytomegalovirus). The results of immunofluorescent staining with IgG, IgA, IgM, C3, and fibrinogen were all negative. Accordingly, a diagnosis of HSP due to acute EBV hepatitis was rendered.
Treatment with intravenous methylprednisolone (1 mg/kg/day three divided doses) and hydration was initiated. Arthralgia, grayish stools, and fatigue disappeared promptly after treatment initiation. The fever subsided on hospital day 2. The swelling on both legs and high GOT/GPT levels improved slowly during the seven days of admission. At discharge, GOT/GPT levels had decreased to 115/274 IU/L, and the T-bilirubin level was normal. Intravenous methylprednisolone administered for one day was switched to oral steroids, which were then tapered off over three weeks. The purpura improved, leaving some dark pigmentation on both legs, but disappeared completely within one month. Periodic urinalyses were performed over the course of one year of follow-up. At two months after discharge, GOT/GPT levels became normal. HSP did not recur and no abnormalities were noted on urinalysis.
Discussion
Hepatic manifestation of EBV infection is commonly transient or self-limited, lasting from a few weeks to several months, but, rarely, can be fatal [3]. Hepatic manifestations include elevated liver enzymes (50-80%), hepatomegaly (10 %), jaundice (5-7%), chronic hepatitis (rare), or fulminant hepatic failure, which is the main cause of death in severe EBV infection [3-5]. No direct EBV cytopathic effect on hepatocytes has been proven [6]. EBV infection of T and B lymphocytes as well as tumor necrosis factor-╬▒ (TNF-╬▒), interferon-╬│, and Fas ligand produced by cytotoxic lymphocytes recruited in the liver have been shown to induce hepatic injury [7].
Skin manifestation occurs in 3-15% of patients with primary EBV infection, including petechiae, purpura, morbilliform rash, erythema nodosum, erythema multiforme, urticaria, and genital ulcer [8, 9]. Mucocutaneous lesions are commonly located on the trunk and upper arms; sometimes they also affect the face and forearms [10]. These symptoms can result from the responses of EBV-specific cytotoxic T lymphocytes to EBV infection, which lead to systemic inflammation [11].
In the 2005, European League Against Rheumatism/Paediatric Rheumatology European Society (EULAR/PRES) modified the classification criteria for HSP as follows: palpable purpura (mandatory criterion) in the presence of at least one of the following four features: 1) diffuse abdominal pain, 2) any biopsy showing predominant IgA deposition, 3) arthritis or arthralgia (acute, any joint), and 4) renal involvement (any hematuria and/or proteinuria) [12]. These criteria support the diagnosis of HSP at the patientŌĆÖs first visit. Acute viral arthritis usually manifests later, rather than in the early acute stage of infection. While granular deposition of IgA and C3 identified by skin biopsy are included in the classic textbook description of HSP, these deposits are not essential diagnostic features [13].
Hypersensitivity vasculitis may be difficult to distinguish from HSP only through skin biopsy. In contrast to patients with hypersensitivity vasculitis, patients with HSP usually have as followings: prominent palpable purpura, arthralgia, abdominal pain, gastrointestinal bleeding, hematuria, normal complement levels, evidence of IgA-containing immune complexes on skin or renal biopsy, and/or an absence of recent use of medications that have the potential to cause vasculitis [14].
HSP can result from exposure to an antigen from the following organisms: Group A streptococcus, parvovirus B19, Bartonella henselae, Helicobacter pylori, Haemophilus parainfluenza, Coxsackie virus, adenovirus, hepatitis A, B, and C viruses, mycoplasma, Epstein-Barr virus, varicella, campylobacter, and methicillin-resistant Staphylococcus aureus. The exact mechanism of HSP development due to primary EBV infection is not known, but contributing factors may include EBV-infected lymphocytes, as well as host responses to the EBV antigen, that lead to the production of various cytokines. EBV has been proposed as a potential initiating factor for autoimmune diseases like systemic lupus erythematosus, rheumatoid arthritis, and primary Sj├ČgrenŌĆÖs syndrome, which is characterized by high titers of anti-EBV antibodies and impaired T-cell immune responses to EBV antigens [15]. In patients with HSP, the level of regulatory T cells (CD4+CD25+) play a major role in preventing autoimmune and inflammatory diseases by secreting anti-inflammatory cytokines, such as interleukin-10 and transforming growth factor beta-╬▓1, the level of which is lower than in normal controls [16]. Furthermore, TNF-╬▒, a potent pro-inflammatory cytokine, is known to play an important role in the occurrence of HSP and HSP nephritis [17].
Kim et al. [18] reported twin cases of HSP nephritis associated with acute EBV infection in south Korea, 2004. Their serological findings were anti-EBV VCA IgM negative, anti-EBV VCA IgG positive, anti-EBV EA IgG positive, and anti-EBV EBNA IgG negative. The differences with our case are as follows: They did not have any symptom of acute EBV infection. Their HSP provoked at least one month after EBV infection by inference from their viral markers. So, compared with our case who presented with HSP accompanying EBV hepatitis, they may be concerned with delayed hypersensitivity reaction in the pathogenesis of HSP.
In summary, we report the case of a 32 month-old girl with acute EBV hepatitis manifesting as HSP. Thus, primary EBV infection could cause HSP.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print