Pathology of C3 Glomerulopathy
Article information

Abstract
C3 glomerulopathy is a renal disorder involving dysregulation of alternative pathway complement activation. In most instances, a membranoproliferative pattern of glomerular injury with a prevalence of C3 deposition is observed by immunofluorescence microscopy. Dense deposit disease (DDD) and C3 glomerulonephritis (C3GN) are subclasses of C3 glomerulopathy that are distinguishable by electron microscopy. Highly electron-dense transformation of glomerular basement membrane is characteristic of DDD. C3GN should be differentiated from post-infectious glomerulonephritis and other immune complex-mediated glomerulonephritides showing C3 deposits.
Introduction
Glomerulonephritides due to alternative complement pathway dysregulation are presently categorized as C3 glomerulopathy [1]. However, the scope of C3 glomerulopathy is somewhat blurred for the following reasons: (i) Some entities, such as atypical hemolytic uremic syndrome (aHUS), may share alternative pathway dysregulation yet differ distinctly in clinicopathologic features [2]; (ii) Various glomerulopathies may show predominance or codominance of C3 deposition (relative to other immunoglobulins or complement types) by immunofluorescence (IF), post-infectious glomerulonephritis (PIGN) being a prime example [3]; and (iii) Laboratory testing for alternative pathway dysregulation is not widely available at present, leaving the current IF standard of measure as the sole determinant of C3 glomerulopathy [4]. Although this subject is complex, the historical aspects of C3 glomerulopathy, its pathogenesis, and pertinent renal biopsy findings are addressed herein.
Historical aspects of disease classification
C3 glomerulopathy is a relatively new disease class established by international consensus in 2013 [5]. Prior to this time, little was known of the related pathophysiology, so many cases previously diagnosed as membranoproliferative glomerulonephritis (MPGN) on morphologic grounds would now qualify as C3 glomerulopathy. Membranoproliferative is a descriptive term, referring to the most common light microscopic finding in such instances and signifying that both glomerular basement membrane (GBM) and related cellular components (mesangial, endothelial, and endocapillary) are duly altered [6].
Repeated past efforts to subclassify MPGN have gradually advanced the understanding and optimal treatment strategies for each subgroup. Despite the impression of homogeneity by light microscopy (LM), three subgroups of MPGN (I–III) emerged, defined by electron microscopy (EM). Subendothelial deposits were characteristic of MPGN type I [7]; and dense deposit disease (DDD), named for its unique electron-dense transformation of GBM, corresponded with MPGN type II [8]. MPGN type III included two distinct forms: the Burkholder variant, showing additional subepithelial immune deposits [9]; and the Strife and Anders variant, demonstrating thickened glomerular capillary walls and destruction of GBM by intramembranous deposits [10,11]. MPGN type II (or DDD) was deemed the most distinctive of these subgroups, given its hallmark EM findings. However, studies conducted during the 2000’s revealed that membranoproliferative patterns by LM were confined to a minority (25–44%) of patients with DDD [12,13], suggesting substantial differences in the pathophysiology of DDD versus other types of MPGN.
In addition to LM and EM features, IF studies have provided critical etiologic clues in this setting fueling the C3 glomerulopathy disease concept. Early investigations from the 1970’s had delineated two distinct IF patterns in instances of MPGN [7,14,15], one of which glomerular deposits of immunoglobulin and C3 both occur and the other is limited to isolated C3 deposition. The former was explained by triggering of complement activation via classical (immune complex-based) or lectin (foreign substance-induced) pathways, whereas isolated C3 deposition was attributed to alternative pathway complement activation.
Fakhouri et al. subsequently introduced C3 glomerulopathy as a new disease class imposed by isolated C3 glomerular deposition (in the absence of immunoglobulin), with or without visible membranoproliferative features [16]. They also defined two subsets of C3 glomerulopathy: DDD bearing dense osmiophilic deposits of mesangium, GBM, and tubular basement membrane and C3 glomerulonephritis (C3GN), a relatively rare and heterogeneous variant with less discrete EM deposits. Although morphologically disparate, DDD and C3GN are similar in etiology and fall within the same disease spectrum [16].
Countless efforts to reach an international consensus on the C3 glomerulopathy paradigm finally bore fruit during the first C3 Glomerulopathy Meeting held in August, 2012 [5]. This gathering of experts in nephrology, renal pathology, complement biology, and complement therapeutics sought to provide a formal definition of the disease and establish guidelines for future research [5]. Unlike the criteria first proposed, they described C3 glomerulopathy as “a disease process due to abnormal control of complement activation, deposition, or degradation and characterized by predominant glomerular C3 fragment deposition with electrondense deposits in EM”. This description implies that small amount of immunoglobulin deposits can be observed in C3 glomerulopathy and that diagnosis of C3 glomerulopathy must rely on IF, LM, EM, and clinical features [5].
Pathophysiology of C3 glomerulopathy
As an important innate defense mechanism against pathogenic stimuli, the complement system includes >30 components and regulators [17]. There are three complement activation pathways: classical, lectin, and alternative. The classical pathway, incorporating C1q, C1r, C1s, C4, and C2 components, is activated by antigen-antibody immune complexes. The lectin pathway, in which C2 and C4 also have roles, is activated by attachment of mannose-binding lectin to microbial carbohydrate groups [17,18]. The alternative pathway is unique in its continual activation by spontaneous C3 hydrolysis, forming C3 convertase (so-called “tick-over” process) [19]. Generation of C3 convertase and its downstream products is tightly regulated by various components of the complement system, so any inherent defects or autoantibody assaults may eventuate in alternative pathway dysregulation [17,20]. The mechanism of alternative pathway dysregulation in the context of C3 glomerulopathy is highly complex, as is regulating the complement system itself. Factors contributing to this dysfunction may be categorized by mechanism of action.
1. Autoantibodies to alternative pathway regulators
C3 nephritic factors (C3NeFs) are IgG or IgM autoantibodies targeting neoepitopes of alternative pathway C3 convertase (C3bBb) [21,22]. C3bBb converts C3 to C3b in conjunction with complement factor B (FB) and complement factor D; and in an amplification loop, factor Bb attached to C3b converts more C3 to C3b [23]. C3 convertase is degraded very quickly by negative regulators, such as decayaccelerating factor (CD55), complement factor H (FH), and complement receptor 1, thus preventing uncontrolled tissue injury. C3NeFs prolong the half-life of C3bBb through interference with these regulators [24-26]. They are also found in many patients with DDD (70–80%) or C3GN (40–55%) [26]. C5 nephritic factors (C5NeFs) are autoantibodies that stabilize C5 convertase (C3bBbC3b) of the alternative pathway, thereby affecting serum C5b-9 levels. Marinozzi et al. have demonstrated C3NeFs in more than half of patients with C3GN, although C3NeFs and C5NeFs in combination or C5NeFs alone may be present; and C5NeFs are more frequently associated with C3GN than with DDD [27]. C4 nephritic factors (C4NeFs) stabilize the C3 convertase (C4bC2a) of classical and lectin pathways. There is evidence of sporadic C4NeF positivity in patients with C3GN, although the recorded prevalence varies [28,29]. The functions of these nephritic factors often require the presence of properdin [17,26,27]. In addition to previous therapeutic trials like plasmapheresis, anti-cellular immune therapy and eculizumab [5], properdin is currently under scrutiny for its therapeutic utility [30]. In addition to nephritic factors, autoantibodies aimed at other regulators or components of the complement pathway (i.e., FH and FB) have been detected [31,32].
2. Genetic alterations
Apart from acquired causes of C3 glomerulopathy thus far discussed, aberrant complement-related genes are similarly implicated, including variants of CFH (affecting FH), CFI (affecting factor I), CFB, and C3 (affecting C3 convertase and C5 convertase) [1,20]. Factor H-related (FHR) proteins are other modulators of the complement pathway, some members (FHR1, FHR2, FHR3, and FHR5) forming dimeric complexes under normal conditions. Abnormal FHR fusion proteins due to genetic alterations may result in C3GN or DDD [1,33]. The above genetic defects cited for C3GN largely overlap with those of aHUS. However, aHUS is marked by solid-phase endothelial cell activation of the alternative complement pathway, unlike the largely fluidphase alternative pathway activation of C3GN [18,34,35]. Alternative pathway dysregulation may also stem from monoclonal immunoglobulin production in patients with monoclonal gammopathy of renal significance, multiple myeloma, and other lymphoproliferative disorders [36,37]. Under such circumstances, monoclonal proteins act as autoantibodies targeting FH or FB and then culminate in C3 glomerulopathy (monoclonal immunoglobulin-associated C3 glomerulopathy) [38].
Pathologic features of C3 glomerulopathy
C3 glomerulopathy is typified by C3 accumulation deposited within glomerular mesangium and capillary walls. Little or no immunoglobulin is present. IF studies confirm an active glomerulonephritis, showing mostly granular or semi-linear C3 deposits more intensely stained (2-level at least) than other complement or immunoglobulin proteins. IgG, IgM, IgA, and C1q deposits may well be present, but not at levels comparable to C3 (Fig. 1A, B), and C4d deposition is rarely seen [5,39].
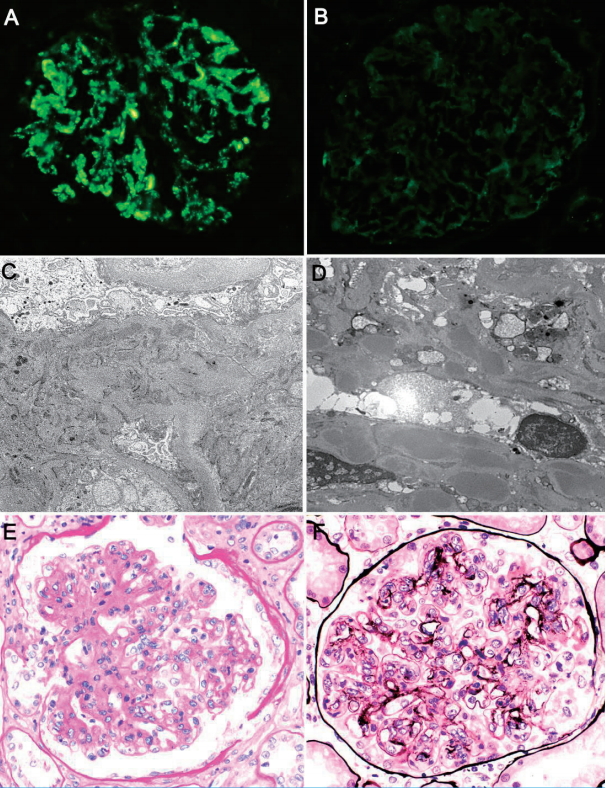
Typical morphologic features of C3 glomerulopathy: Immunofluorescence preparations show (A) strong granular staining of glomerular C3 deposits (within mesangium and along capillary walls) as well as (B) trace IgG. Electron microscopic examination confirms multiple electron-dense deposits of (C) mesangium (×6,000) and (D) subendothelial regions (×7,000). Light microscopy displays (E) mesangial proliferation (Periodic acid-Schiff, ×400) and (F) double-contoured capillary walls (methenamine silver, ×400), indicating membranoproliferative glomerulonephritis.
Electron-dense glomerular deposits are identified in all forms of C3 glomerulopathy and serve to differentiate the two major subtypes of C3 glomerulopathy: DDD and C3GN [5]. DDD is defined by highly electron-dense and osmiophilic intramembranous deposits and resultant GBM lamina densa transformation. The deposits appear ribbon- or sausage-like and may even inhabit tubular basement membrane or Bowman’s capsule. In patients with C3GN, these deposits are ill-defined and not as highly electron-dense, involving mesangium and subendothelial regions (Fig. 1C, D). Rarely are they subepithelial or intramembranous in nature. They resemble those commonly seen in immune complex-mediated glomerulonephritis. So-called “hump-like” subepithelial deposits, similar to those manifested in PIGN, may be identified at times.
LM findings of C3 glomerulopathy are diverse, ranging from near-normal morphology to features of proliferative (mesangial, membranous, or endocapillary), crescentic, or sclerosing glomerulonephritis. The pattern witnessed in C3GN is usually that of MPGN (Fig. 1E, F) [40-43], characterized by mesangial expansion with hypercellularity and thickened (i.e., doubly contoured) glomerular capillary walls. In one series, MPGN patterns accounted for 71% of patients with C3GN [42]. The others primarily showed mesangial proliferation, without changes in glomerular capillaries. Some will lack mesangial proliferation altogether, mimicking minimal change disease. The glomeruli may show endocapillary proliferation, with variable inflammatory cell infiltrates, and crescents may be encountered [5,42,44].
In a large series of patients with DDD, the most common histologic pattern was mesangial proliferative GN (43.4%), followed by membranoproliferative (24.6%), crescentic (17.4%), and endocapillary proliferative (11.6%) glomerulonephritis [13]. Other cohorts have most often displayed features of MPGN (43.8–77.8%), followed by mesangial proliferative glomerulonephritis [12,45,46].
Differential diagnosis of C3 glomerulopathy
1. C3 glomerulonephritis versus dense deposit disease
DDD is sometimes difficult to distinguish from C3GN [5,47]. As noted earlier, C3 glomerulopathy in the absence of highly electron-dense, ribbon-like deposits by EM is classifiable as C3GN; and the electron-dense deposits of C3GN are more often found in mesangial and subendothelial regions, seldom occupying the intramembranous and subepithelial areas typical of DDD [48]. Assessments of deposit density are subjective, frequently sparking disagreement among pathologists in their classification of DDD vs C3GN [5,47,49-52]. Furthermore, the ribbon-like deposits of DDD are generally discontinuous, creating the potential for sampling error and subsequent misdiagnosis as C3GN. There are no other specific LM criteria for differentiating between DDD and C3GN, except when intramembranous deposits stain intensely by Periodic acid-Schiff [8,53]. Such deposits are fuchsinophilic in trichrome stains and lack methenamine silver positivity [53].
The ratio of DDD to C3GN is reportedly about 1:3 [42,54]. It may be necessary to distinguish between DDD and C3GN for prognostic or therapeutic studies that compare respective outcomes. There is greater likelihood of C3NeFs [42] and lower circulating C3 levels in patients with DDD (vs C3GN) [54]. Otherwise, no clear clinical differences are evident.
2. Immune complex-mediated glomerulonephritis
By definition, glomerular C3 deposition in patients with C3 glomerulopathy is at least 2-level more intensely stained in IF preparations than are other complement or immunoglobulin deposits. However, the distinction between C3 glomerulopathy and MPGN is not always obvious on this basis. In terms of the latter, glomerular staining of C4d (a byproduct of activated classical and lectin pathways) may be helpful [55]. C4d staining is negative or trace in instances of C3 glomerulopathy, whereas immune complex-mediated glomerulonephritis yields positive results [55-57].
3. Post-infectious glomerulonephritis
PIGN is a self-limited glomerulonephritis commonly triggered by streptococcal infection [58] and reflecting a bacterial immunologic response. The findings are those of immune complex-mediated disease. Diffuse endocapillary glomerulonephritis, deposition of IgG and C3, and subendothelial and subepithelial hump-like deposits are usually visible by LM, IF, and EM, respectively.
Difficulty may arise in distinguishing some cases of PIGN from C3GN. C3 amplification may outlast immunoglobulin, and C3-predominant or isolated C3 deposits may exist [59]. C3GN may then sporadically present as a diffuse endocapillary proliferative pattern [5,48,54,60], and hump-like subepithelial deposits are frequent accompaniments of both MPGN and C3GN [48]. The clinical course and laboratory findings are perhaps helpful in this regard. Patients with PIGN readily recover baseline kidney function and laboratory profiles in a matter of weeks, resolving hematuria, proteinuria, and hypocomplementemia without therapeutic intervention. Prolonged proteinuria and low serum C3 levels are clinically indicative of chronic glomerulonephritis. However, initial PIGN presentations have seemingly morphed into C3 glomerulopathy [57,61,62], transformed (it now appears) by complement activation via alternative pathway [61-64]. Various sources have also noted the presence of nephritis-associated plasmin receptor (NAPlr) in instances of PIGN with C3 glomerulopathy features [63,65,66]. As shown by Sethi et al., most cases of PIGN plagued by persistent hematuria and proteinuria have involved underlying defects, either genetic mutations, autoantibodies, or both, that impact alternative complement pathway regulation [57]. Before conceding a diagnosis of C3 glomerulopathy in patients with C3-predominant PIGN, persistent hypocomplementemia, and proteinuria or declining renal function, further investigation of the alternative complement pathway is consequently advised [3,5].
Conclusion
C3 glomerulopathy is a recently established disease entity pathogenetically linked to alternative complement pathway dysregulation. The histologic patterns entailed are diverse, membranoproliferative being the most common. Based on distinctive EM features, DDD and C3GN are specific subtypes of C3 glomerulopathy, both requiring differentiation from other glomerulopathies marked by a predominance of C3 deposition.
Notes
Conflict of interest
The authors declare that they have no conflicts of interest.