Nonobstructive Bilateral Hydronephrosis & Hydroureter from Nephrogenic Diabetes Insipidus with a Novel Mutation of AQP2 Gene (p.A123G)
Article information
Abstract
Nephrogenic diabetes insipidus (NDI) can cause nonobstructive hydronephrosis. Congenital NDI (CNDI) is caused by a genetic mutation. This case report presents a 12-year-old girl who was incidentally diagnosed with nonobstructive hydronephrosis due to NDI caused by AQP2 gene mutation after being evaluated for microscopic hematuria found on routine health examination at school. The patient’s medical and family history was unremarkable, and she complained of nocturia only at the time of the clinic visit. Bilateral hydronephrosis on abdominal ultrasonography prompted a water deprivation test, leading to diagnosis of NDI. Genetic study confirmed p.Asn (AAC)123Ser (AGC) in exon 2 of the AQP2 gene. Polyuria and hydronephrosis improved following arginine-vasopressin therapy. CNDI responsive to treatment should be considered as a possible cause of nonobstructive hydroureter.
Introduction
Nonobstructive hydronephrosis may occur in the absence of anatomic urinary tract obstruction when urinary output exceeds the capacity of the genitourinary system. Major causes include diseases that increase urine production, such as nephrogenic diabetes insipidus, central diabetes insipidus, and primary polydipsia [1,2]. Persistent urine volume overload obstructs the urine flow and results in reflux, and, therefore, warrants appropriate volume control in such cases [3].
NDI can occur sporadically- lithium being the most well-known medication to cause the disease- and can also occur congenitally due to genetic defect [1]. 90% of genetic mutation associated with NDI is X-linked AVP gene mutation, with the remaining 10% being mutation of autosomal recessive AQP2 gene [4]. NDI caused by genetic mutation may also respond to AVP therapy if the patient has heterozygous mutation [5]. Therefore, the identification of the cause of NDI and appropriate treatment targeting the underlying problem is critical. We report a case of a 12-year-old girl with incidentally discovered bilateral nonobstructive hydronephrosis & hydroureter from CNDI with a novel mutation of AQP2 gene (p.A123G).
Case report
A 12-year-old girl was referred to the Department of Pediatrics of Wonju Severance Christian Hospital for microscopic hematuria found on routine clinical examination performed 1 year ago. The patient complained of nocturia, and she had a history of undergoing surgery for ventricular septal defect. Medical history of the patient’s mother was unremarkable.
On physical exam, the patient presented with clear breath sounds and regular heartbeats. Her body weight was 33.0 kg (3.5th percentile) and height 144.0 cm (5th percentile). Systolic and diastolic blood pressure were 98 mmHg (103 mmHg being 50th percentile) and 61 mmHg (59 mmHg being 50th percentile), respectively.
Laboratory studies were as follows: serum sodium 145 mmol/L, potassium 4 mmol/L, chloride 108 mmol/L, carbon dioxide 27.8 mmol/L, blood urea nitrogen 7.5 mg/dL, creatinine 0.54 mg/dL (calculated effective glomerular filtration rate 110.13 mL/min/1.73m2), calcium 9.0 mg/dL, phosphorous 5.19 mg/dL and magnesium 2.2 mg/dL. Serum osmolality and urine osmolality was 296 mmol/kg and 80 mmol/kg, respectively. Urinalysis was as follows: sodium <5 mmol/L, potassium 5.3 mmol/L, chloride <50 mmol/L, carbon dioxide 10.4 mmol/L, creatinine <1.0 mg/dL, calcium 0.01 mg/dL, total protein 1.5 mg/dL, microalbumin 0.2 mg/dL and cystatin C 0.01 mg/dL. Hormonal assays were measured as well, with serum antidiuretic hormone (AVP) level being 5.21 pg/mL (reference range: <6.7 pg/mL) and parathyroid hormone (PTH) being 92.43 pg/mL (reference range: 15 to 65 pg/mL). Serum and urine osmolality measured after overnight fasting were respectively 309 mmol/kg and 97 mmol/kg, which prompted the diagnosis of diabetes insipidus.
Abdominal sonography (US abd) and intravenous pyelogram (IVP) were performed, which found bilateral hydronephrosis and hydroureter (Fig. 1). There was no detectable reflux on voiding cystouretherogram (VCUG), and there was no remarkable obstructive lesion on diuretic renogram (Fig. 2).

IVP images before and after AVP treatment. (A) IVP image before AVP treatment. There are both hydronephrosis and hydroureter. (B) IVP image after AVP treatment. Both hydronephrosis and hydroureter are resolved.
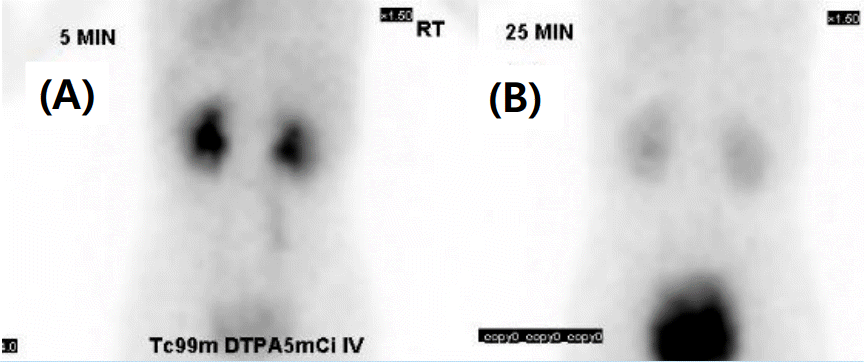
Diuretic renogram images. (A) Diuretic renogram before diuretics injection (B) 25 min later image(diuretics injection at 17 min later). There is no obstructive lesion.
The patient was tested for genetic mutations associated with NDI, and heterozygous A123G missense of AQP2 gene was found, while that of the patient’s mother was normal (Fig. 3).
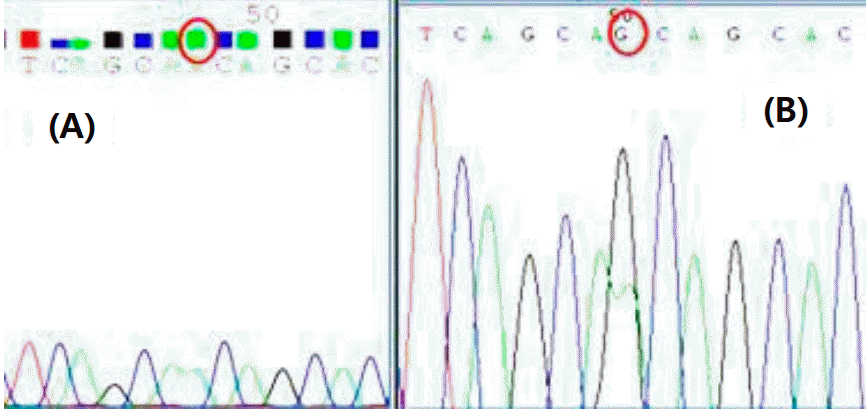
Result of sequencing of exon2 of the AQP2 gene. (A) Normal DNA sequence (B) Sequence of DNA from patient, who was heterozygous for the A to G mutation.
After the diagnosis, the patient underwent a trial of thiazide therapy to which she was unresponsive. However, she was responsive to AVP therapy, and a further diagnosis of partial NDI was made. After AVP therapy for 7 weeks, her serum and urine osmolality improved to 275 mmol/L and 713 mmol/L, respectively, with urine specific gravity exceeding 1.030. The patient’s polydipsia and polyuria also improved. Improvements of hydroureter and hydronephrosis were also noted on follow-up IVP and abdominal ultrasonography, which completely resolved 18 months later (Fig. 1). At the age of 16, 4 years after starting AVP therapy, the patient’s body weight is 49.8 kg (between 25 and 50th percentile) from 30.0 kg (3rd percentile) and her height is 157.7 cm (25th percentile) from 144.0 cm (5th percentile), demonstrating improvement in growth pattern as well.
Discussion
The patient in this case report was diagnosed with NDI, specifically CNDI resulting from genetic disorder. Mutations in AVPR gene or AQP2 gene can cause CNDI, with the former being far more common [4]. Fujimoto M et al. [6] reported only 8 patients (5.59%) out of 143 CNDI patients to have AQP2 gene mutation. The genetic mutation found in this patient (exon2 p.A123G of AQP2 gene) was not found on Ensembl, a well-known genetic browser. This is the first report of the incidence.
CNDI is generally diagnosed in infantile stage. Typically, the infant developed intermittent fever and hypernatremia at one month of age [6,7]. However, this patient was mostly asymptomatic except for nocturia, and further evaluation was made only after an incidental finding of microscopic hematuria on annual health checkup. The diagnosis of diabetes insipidus was made after imaging studies found nonobstructive hydronephrosis and hydroureter.
In most cases of nonobstructive hydronephrosis and hydroureter, long-standing volume overload causes bladder distension, and detrusor hypertrophy of the bladder neck develops as part of compensation process, resulting in obstruction and urinary reflux [3,8,9]. Several case reports of nonobstructive urinary tract dilation due to underlying complete NDI leading to cystostomy formation have been made as well [10].
Despite sharing common genetic mutation, homozygous mutation shows larger urinary output and is less responsive to treatment [5]. In contrast, this patient’s mutation was heterozygous, which may explain the milder course of disease. Urinary tract dilation and growth restriction improved following AVP therapy.
This patient’s case suggests that the diagnosis of NDI, which is treatable, should be a part of a list of differential diagnosis in children presenting with nonobstructive hydroureter.
Notes
Conflict of interest
No potential conflict of interest relevant to this article was reported.