Introduction
Bartter syndrome is a rare hereditary hypokalemic tubulopathy. It presents with two broad phenotypes, one with antenatal Bartter syndrome, and the other with classic Bartter syndrome, typically associated with mutations in the CLCNKB gene [1]. While all forms of Bartter syndrome are characterized by hypokalemic, hypochloremic, and hyperreninemic metabolic alkalosis with normal blood pressure, the clinical presentation of classic Bartter syndrome is very heterogeneous, ranging from very mild to severe [2]. Here, we report a Russian 6-year-old identical twin who presented with severe hypokalemia and multiple renal cysts, and they were found to have Bartter syndrome with a novel homozygous c.1614delC deletion on exon 15 of the CLCNKB gene .
Case report
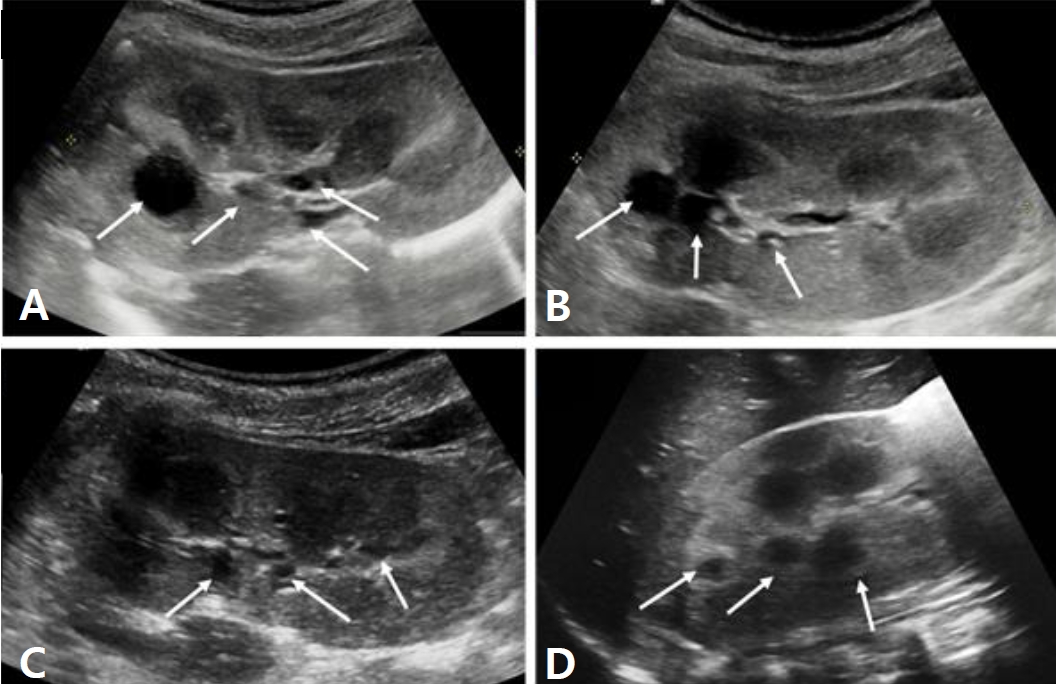
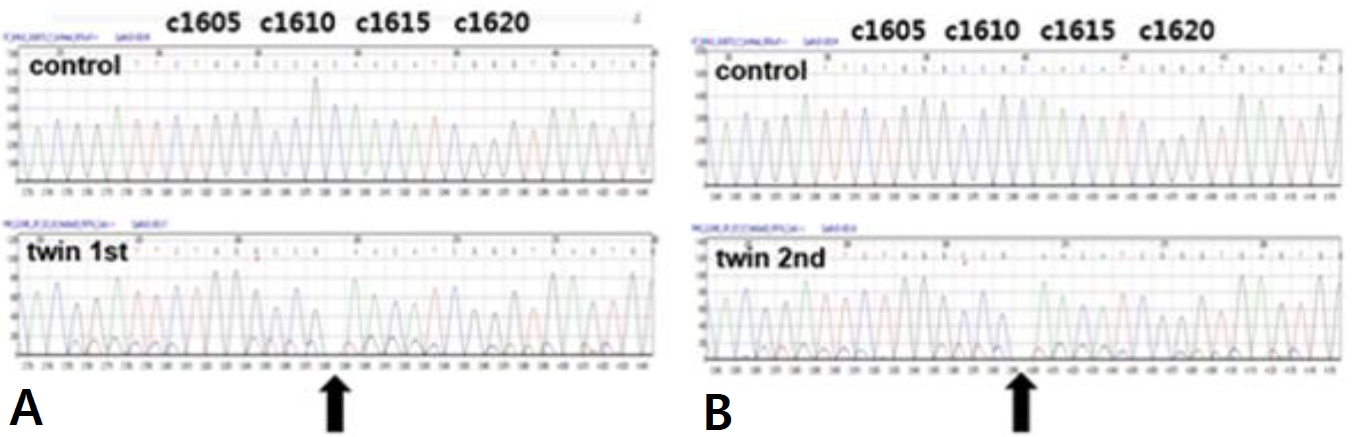
A six-year-old male identical twin from Russia visited ou r hospital for the evaluation of renal cysts. Kidney cysts were incidentally found throughout the evaluation of acute diarrhea in Russia 6 months ago. The twin boys had no specific pre-natal and perinatal histories. At 18 months of ages, they were evaluated for short stature in Russia, which showed no specific findings. Since two years of age, both boys developed polydipsia, polyuria, and nocturia. They were not on any medications, and their family history was unremarkable. The first twinŌĆÖs height was in the 7th percentile and body weight was in 16th percentile. His blood pressure levels were normal. There were no specific findings on physical examination. The laboratory findings for the first twin showed metabolic alkalosis (pH 7.545 and HCO<sub>3</sub> ŌĆō 31.6 mmol/L), hyponatremia (131 mmol/L), severe hypokalemia (2.3 mmol/L), hypochloremia (81 mmol/L), and normal serum magnesium level (2.1 mg/dL). Transtubular potassium gradient (TTKG, 12.2) and fractional excretion of chloride (2.56%) were elevated. Urinary calcium excretion was normal. Renin was elevated (30.48 ng/mL/ hr, normal 0.8ŌĆō2.0 ng/mL/hr), but aldosterone was normal (26.4 ng/dL, normal Ōēż40 ng/dL). Urine ╬▓2-microglobulin was increased (2.57 ╬╝g/mL, normal 0ŌĆō0.3 ╬╝g/mL). The second twin also had hyponatremia (133 mmol/L), hypokalemia (2.5 mmol/L), and hypochloremia (83 mmol/L). The total CO2 level was 29.8 mmol/L. The levels of magnesium were within the normal range. The renal potassium and chloride wasting was also found with TTKG of 14.4 and urine chloride of 43 mmol/L. Renin (30.48 ng/mL/hr) level was elevated and aldosterone (26.4 ng/dL) was normal. Electrocardiogram showed mild corrected QT prolongation, however, echocardiographic findings showed normal findings. Renal sonography of both boys showed nephromegaly with increased cortical echogenicity and multiple cysts of varying sizes in both kidneys (Fig. 1A-D). Although cystic kidney diseases such as autosomal recessive polycystic kidney disease (ARPKD) were initially suspected on renal sonography, their laboratory and clinical findings were consistent with Bartter syndrome. There were no specific findings compatible with ARPKD. Therefore, they were diagnosed with Bartter syndrome with renal cysts. Due to financial problem, only the first twin was admitted to the hospital for the treatment of severe hypokalemia. After admission, serum potassium and chloride levels did not increase effectively, despite vigorous potassium (5.0 mEq/kg/d) and chloride (7.09 mEq/kg/d) supplementation. Renal potassium wasting and polyuria persisted (urine volume 3.11 L/m2/day). Non-steroidal anti-inflammatory drug (ibuprofen) and angiotensin-converting enzyme inhibitor (lenipril, 0.13 mg/kg/day) were added from the third day of admission. The hourly urine output was reduced from 9.0 mL/kg/hr to 3.09 mL/kg/hr following the administration of ibuprofen (Fig. 2A). The boy was discharged with mildly improved metabolic alkalosis, hypokalemia and hypochloremia on the fourth hospital day (Fig. 2B). Potassium chloride (K-contin, 2.56 mEq/kg/day), lenipril (0.13 mg/kg/day), and ibuprofen were prescribed. The second twin who was treated through an outpatient clinic was also prescribed potassium chloride (K-contin, 3.08 mEq/kg/day), lenipril (0.1 mg/kg/day), and ibuprofen. They returned to Russia with a 30-day medication and a referral form. The genetic analysis for both boys revealed a homozygous c.1614delC deletion on exon 15 of the CLCNKB gene, which is a previously unreported variant to the best of our knowledge (Fig. 3A and B). CLCNKB gene encodes the kidney-specific chloride channel CLC-Kb.
Discussion
This is a case report of classic Bartter syndrome in an identical twin who presented with renal cysts, polyuria, polydipsia, and hypokalemic, hypochloremic and hyperreninemic metabolic alkalosis with normal blood pressure. While ARPKD was suspected initially, their clinical and laboratory findings excluded ARPKD but suggested Bartter syndrome. The genetic analysis for both boys confirmed a novel homozygous mutation of the CLCNKB gene.
Bartter syndrome is a rare AR salt-losing tubulopathy, identified by Frederic Bartter in 1962 [3]. It is clinically categorized into antenatal or classic Bartter syndrome, and five genetic subtypes with mutations in SLC12A1, KCNJ1, CLCNKB, BSND, and CASR have been identified to date [4]. These are all autosomal recessive types, but recently X-linked inheritance-type bartter syndrome was revealed, which is mutations in the gene encoding the protein melanoma associated antigen D2 (MAGE-D2) [5]. Classic Bartter syndrome is caused by the mutation in CLCNKB in the chloride channel family of genes. Its features for older children include failure to thrive, dizziness, polyuria, polydipsia, non-specific fatigue and muscle weakness [1]. To the best of our knowledge, there have been no cases of classic Bartter syndrome presenting with multiple renal cysts in identical twins, as in our case. The twins showed previously unreported mutation on exon 15 of the CLCNKB gene.
Notably, renal cysts are frequent findings in patients with chronic hypokalemia owing to hyperaldosteronism or other renal potassium-wasting disorders [6,7]. In a study analyzing the 55 patients with hyperaldosteronism [6], 44.4% had renal cysts. Lower serum potassium levels and higher plasma renin activity independently predicted the presence of kidney cysts. In addition, the number and size of renal cysts were reduced strikingly after adrenal adenomas were removed, which was consistent with the normalization of the serum potassium levels. Meanwhile, renal cyst formation is rarely reported in Bartter syndrome, but, in 2005, a case of bilateral renal cysts and nephrocalcinosis was described in a 6-year-old girl with classic Bartter syndrome [8]. She had a heterozygous nonsense mutation (W610X) on exon 16 of the CLCNKB gene. While nephrocalcinosis is almost invariable in antenatal Bartter syndrome, the authors suggested that nephrocalcinosis may develop in patients with classic Bartter syndrome and that both hypokalemia and nephrocalcinosis might contribute to the development of renal cysts [8]. Our twin case did not show any evidence of nephrocalcinosis, but they had various sizes of multiple renal cysts and mutation of the CLCNKB gene. Persistent severe hypokalemia seemed to be associated with the presence of renal cysts in this clinical setting.
Renal cysts may develop as a complication of deregulated solute transport and increased renal cell proliferation under the condition of chronic hypokalemia. Decreased extracellular potassium concentration enhanced renal cell proliferation in vitro and collecting ductal hyperplasia was observed in potassium-deficient rats [9,10]. Furthermore, hypokalemia stimulates glutamine uptake and ammoniagenic enzyme expression in the proximal tubule [11]. Increased renal ammonia production can stimulate DNA, RNA and protein synthesis, facilitate renal cell growth, and increase interstitial injury during which renal cyst formation may increase [11]. In both patients, the renal sonography showed nephromegaly, increased parenchymal echogenicity, and multiple various sized cysts. Medical history, clinical presentations, and laboratory findings were sufficient for ruling out any possibility of PCKD. The urinary ╬▓2-microglobulin level was also elevated, suggesting tubular injury. These findings indicate that persistent severe hypokalemia may play a role in renal tubular cell hypertrophy, tubulointerstitial injury, and cyst formation. Another point to consider is that the renin angiotensin aldosterone system (RAAS) activation can promote the growth of renal cysts through several signaling cascades. Angiotensin II is known to promote vascular hyperplasia, cyst expansion, and intrarenal ischemia, leading to the disruption of the renal architecture [12]. Lastly, the mutation of the CLCNKB gene in our cases could fundamentally contribute to the renal cystic growth. Chloride channels plays an essential role for cell volume regulation and transepithelial transport, and the novel homozygous deletion on exon 15 of the CLCNKB gene in our identical twin cases would cause more severe loss of function than seen with other known mutations. The wide spectrum of functional severity of CLCNKB mutations correlated with the phenotypic variability in patients with classic Bartter syndrome [13]. Since functional analyses were performed in only 20 of the CLCNKB mutations [2], further investigation for the impact of CLCNKB mutations on the chloride channel activity is necessary.
In patients with Bartter syndrome, comprehensive therapy such as potassium supplementation, prostaglandin inhibitors, RAAS blockers and potassium-sparing diuretic is required [4]. Our patients were treated with potassium chloride, lenipril, and ibuprofen. Metabolic alkalosis, hypokalemia, hypochloremia, and polyuria partially improved during the short course of treatment. Because this family did not revisit our clinic after leaving our country, we could not perform any genetic investigation for the parents. Therefore, we do not clearly confirm whether the mutation was de novo or inherited from their parents. Although Bartter syndrome rarely progresses to end-stage renal disease, chronic kidney disease with severe proteinuria developed in a substantial number of patients during follow-up [14,15]. To manage Bartter syndrome in a timely manner, we should carefully take medical history, promptly make a diagnosis, appropriately treat electrolyte imbalance, and finally improve their quality of life.
Conclusion
This is the first report of a novel homozygous mutation in the CLCNKB gene in an identical twin, presenting with multiple renal cysts. Although the mechanisms underlying renal cyst formation remain unclear, mutations in the CLCNKB gene may reveal variable Bartter phenotypes. Functional study of the mutations and long-term followup are necessary to provide targets for individual treatment in patients with Bartter syndrome.

 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print