Introduction
Chronic kidney disease (CKD) in children is defined as the presence of structural and functional abnormalities or a decline in glomerular filtration rate for more than 3 months, and can progress to end-stage renal disease (ESRD) [1]. The prognosis of CKD has improved remarkably with improved medical treatment, dialysis techniques, and transplantation. However, CKD is still associated with growth impairment, lower quality of life, cardiovascular morbidity, and increased risk of mortality in the pediatric population [2-4]. The main etiologies of pediatric CKD are congenital anomalies of the kidneys and urinary tract (CAKUT), steroid-resistant nephrotic syndrome (SRNS), chronic glomerulonephritis, and renal cystic ciliopathies, which account for more than 70% of pediatric CKD altogether [5-7].
A positive family history of kidney disease is reported in approximately 25 % of patients with kidney disease, suggesting the importance of genetics in kidney disease [8,9].With recent advances in genetic testing technologies, genetic testing has become a mainstay in clinical diagnostics and research. As a result, knowledge about genes associated with kidney and urological disorders continue to expand, and more than 600 genetic disorders have been identified in kidney disease [10,11]. Genetic kidney disease is more prevalent in pediatric patients than in adult patients, and is estimated to account for approximately 30% of pediatric chronic kidney disease [10].
From the perspective of this genetic heterogeneity, selection of appropriate genetic testing is not an easy decision, and is generally made based on the patientsâ clinical impression, the diagnostic yield of a test, and its cost. In this review, we introduce various genetic testing methods and describe the characteristics of each modality to help pediatric nephrologists select the appropriate genetic testing method as part of the genetic diagnostic process in children with kidney disease.
Diagnostic genetic testing and diagnostic yield
The human genome contains approximately 20,000 genes, with approximately 1% to 2% being protein-encoding exonic regions, known as the exome. Another 26% form intronic regions between exons, and the remaining 72% to 73% of intragenic regions form noncoding regions [12]. Genetic variations range in size from single nucleotide variants (SNVs) that substitute a single base and small insertion/ deletion (INDEL) mutations that affects 2â1,000 bases, to large copy number variations (CNVs) that affect more than 1,000 bases.
Genetic tests available in clinical practice include karyotyping, chromosomal microarray (CMA), Sanger sequencing, targeted gene panel, whole exome sequencing (WES), and whole genome sequencing (WGS). Table 1 summarizes the characteristics of the different genomic diagnostic modalities used.
Karyotyping directly visualizes the entire set of 46 chromosomes during prometaphase or metaphase with a high sensitivity for aneuploidies, large deletions and duplications, and translocations. CMA is more sensitive for the detection of microdeletions or microduplications that are over 20â400 kilobases (Kb) in size, but cannot detect balanced translocations or inversions [13-15]. CMA in combination with karyotype is the test of choice when a structural variant is clinically suspected, as in developmental delay and multiple congenital anomalies. Recent studies have reported a high frequency of CNVs in CAKUT, involving 45 different genomic disorders at 37 independent loci, with six of these loci (1q12.1, 4p16.1â16.3, 16p11.2, 16p13.11, 17q12, 22q11.2) accounting for approximately 65% of cases with a genomic disorder [16]. Structural variants have been detected in approximately 10%â17% of children with CAKUT [17,18].
Sanger sequencing, a targeted dideoxy terminator sequencing, identifies small variants of less than 1 kb in specific genes or gene regions. It is simple with highly reliable results having a low error rate (0.001%â1%), and has been considered the gold standard for accurate genetic diagnosis [19]. Sanger sequencing is used for testing when a variation is clinically suspected at a specific locus.
Next-generation sequencing (NSG), also known as massive parallel sequencing, uses simultaneous sequencing of millions of small DNA fragments that can be reassembled into a full sequence using analytical tools. This approach is a powerful tool for identifying small variants in the whole exome, whole genome, or selected genes at a relatively low cost. It has emerged as a powerful tool for clinical genetic diagnosis [20].
The targeted gene panel covers a selected set of genes that are considered to be associated with a specific phenotype. It is a rapid and cost-effective test for a clearly distinct phenotype with several causative genes, such as nephronophthisis, Alportâs syndrome, SRNS, and focal segmental glomerulosclerosis (FSGS) (Table 2) [21-24]. The diagnostic yield depends on the clinical diagnosis and selection criteria. In recent reports, the causative mutations were found in 8%â20% of children with CAKUT, approximately 26% of children with SRNS, and up to 80% of patients with cystic kidney disease [10,25-28]. This targeted approach simplifies the interpretation of the results, minimizes potential false negative results caused by incomplete sequencing coverage, and reduces the opportunity for incidental findings. However, because the genes in a particular panel are selected, it is not possible to reanalyze the data panels when new or phenocopy genes are discovered. For example, isolated proteinuria or SRNS may be the only evident clinical sign of Alport syndrome, Dent disease, or Fabry disease at disease onset or even later. These genetic kidney diseases are often misclassified as SRNS, and the results of targeted genetic testing prove to be negative. These conditions are referred to as the phenocopies of hereditary SRNS [29,30].
WES and WGS are relatively unbiased genome-wide tests. WES covers protein-coding regions of the human genome, whereas WGS covers all genomic DNA, including protein-coding and noncoding regions. WES is less expensive, detects a reduced number of variants compared to WGS, and approximately 75% of variants are located within the exome. On the other hand, WGS is efficient for detecting structural variants and variants in splicing or regulatory regions [31,32]. WES and WGS allow for future reanalysis without the need for additional sequencing, as our knowledge on the function of a gene and analytic techniques improves with the discovery of a new causative gene [33].
In general, WES and WGS yield higher diagnostic results than targeted gene panels [34-37]. The diagnostic rate of genetic forms of kidney diseases was higher among patients with a positive family history of kidney disease, consanguinity, congenital forms of kidney disease or early age of onset, and extra-renal manifestations [30,35,38].
Nonetheless, NGS coverage is suboptimal in genes related to kidney disease, such as PKD1 and GREB1L [39,40]. In addition, tandem repeats, which are short repetitive nucleotide sequences, are difficult to sequence with short-read NSG technology.
The large amount of sequence variations and incidental findings raise the issue of interpretation in NGS. The American College of Medical Genetics and Genomics (ACMG) recommends that variants be classified into one of five categories: pathogenic, likely pathogenic, variant of uncertain significance (VUS), likely benign, and benign. The variant is assessed using various types of evidence, including population frequency, in silico prediction, functional study data, segregation data, de novo mutation, variant characterization, and prior occurrence [41]. The terms âpathogenicâ or âbenignâ are used for variants of almost certainty, and âlikely pathogenicâ and âlikely benignâ for those of over 90% certainty. The variants that have insufficient evidence to be classified into the above categories are reported as VUS.
There are concerns regarding the interpretation of the indeterminate results reported as VUS. Without an understanding of the variation of the human genome, this result can be misinterpreted as significant. Importantly, recent reports indicate that many VUS cases can be reclassified as benign [42,43].
Regarding the possibility of incidental findings, ACMG lists 59 medically actionable genes, including those of cancer and cardiomyopathy, to be assessed in patients undergoing WES or WGS [44]. There are ethical issues around incidental findings, particularly in children, since the advantages of informing children or their family of incidental findings related to adult-onset diseases is unclear. The American Academy of Pediatrics recommends delaying testing for adult-onset disease until adulthood [45].
Clinical implications of genetic diagnosis
Genetic testing in patients with kidney disease has several clinical implications which can contribute to a definitive diagnosis. Establishing an accurate diagnosis is necessary for the effective management of kidney disease based on the pathophysiology of the disease. For example, avoidance of corticosteroid and CoQ10 supplementation can be considered in patients with SRNS caused by defects in genes of the CoQ10 biosynthesis pathway [46].
In addition, genetic diagnosis provides a better understanding of long-term prognosis and enables early evaluation, referrals, and follow-up of extrarenal manifestations of syndromic disease. CAKUT caused by HNF1B mutations or 17q12 microdeletion is related to hypomagnesemia, hyperuricemia, and maturity-onset diabetes of the young (MODY) type 5. Similarly, PAX2 , EYA1, and SALL1 have been associated with CAKUT and deafness, GATA3 with CAKUT and hypoparathyroidism, WT1 with nephrotic syndrome and increased risk of Wilmsâ tumor and gonadoblastoma; and NPHP5 with nephronophthisis and retinitis pigmentosa, resulting in progressive blindness [47-52]. Thus, early detection of these conditions can lead to early treatment.
Genetic diagnosis enables avoidance of unnecessary or invasive diagnostic procedures such as kidney biopsy for the diagnosis of Alportâs syndrome in the proband or in at-risk relatives. Genetic diagnosis also enables precise genetic counseling, identification and testing of at-risk relatives, prediction of disease recurrence for future pregnancies, and preimplantation and prenatal diagnosis.
Genetic counseling
When genetic testing is required, appropriate genetic counseling, including documentation of consent from the patient, is essential before testing [53]. This discussion includes an explanation on the basic principles of heredity, genetic disease, and testing modality; the benefits, risks and limitations of testing; expected testing results; types of incidental/secondary findings; and cost of testing. Patients should be informed of the possibility of incidentally finding medically actionable genes.
Genetic counseling should be provided after testing. The test results should be interpreted, and the inheritance pattern and risk of recurrence need to be discussed. In addition, the availability of management and prevention modalities, the need for further testing, and the implications for the family should be discussed. Moreover, emotional and social support and patient support groups or reference resources should be provided.
Diagnostic approach and interpretation of genetic testing
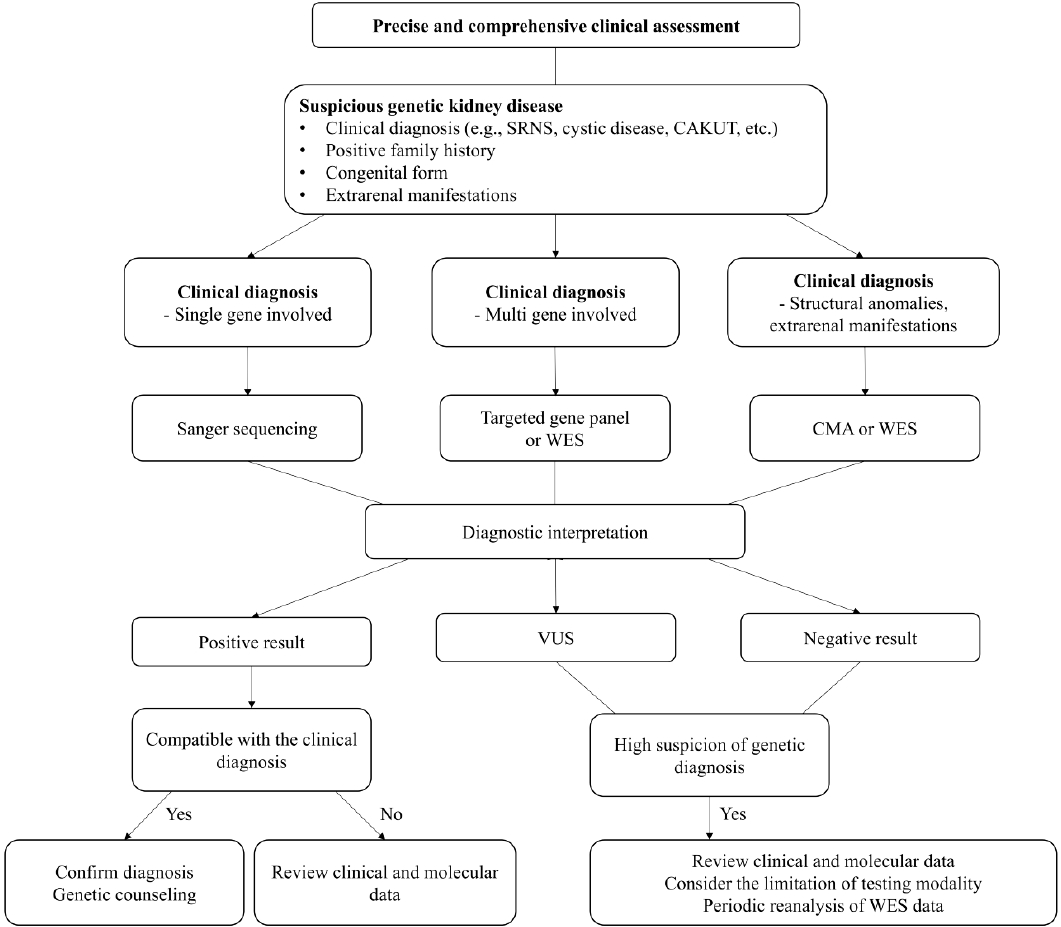
The most appropriate genetic testing modality should be determined according to the clinical diagnosis. The Fig. 1 shows the diagnostic approach for genetic testing of kidney disease, modified from the suggested algorithm for kidney disease of unknown etiology by Hay et al. [15]. When genetic testing results are positive, we should check whether the genetic diagnosis is consistent with the patientâs clinical manifestations. The strategy of re-evaluation of patients after genetic testing, i.e., reverse phenotyping, increases the diagnostic yield and enables the identification of subtle and previously unrecognized clinical signs [29,54].
If the genetic diagnosis is compatible with the clinical diagnosis, appropriate genetic counseling should be performed with patients and at-risk relatives. If the genetic test result is incompatible with clinical manifestations, we should review the clinical and molecular data, and discuss it with molecular diagnosticians or clinical geneticists.
There should be high suspicion for genetic diagnoses despite negative genetic testing results, considering the limitations of genetic testing modalities. For example, WES has blind spots and cannot detect deep intronic splicing variants, large deletions, or duplications. Additionally, somatic mosaic mutations that are not detected in DNA extracted from peripheral leukocytes may be present.
Conclusion
The role of genetic testing in the diagnosis of kidney disease has increased in recent years. It is important for pediatric nephrologists to know when and how to apply genetic testing modalities. Genetic testing is recommended in children with SRNS, in those suspected hereditary forms of kidney disease such as CAKUT or cystic disease, and in those with extrarenal manifestations [55,56]. Genetic testing alone, without a detailed phenotype, rarely yields a confirmative diagnosis. Therefore, careful, precise, and comprehensive assessment by clinicians is essential, encompassing patient history, family history, physical examination, biochemical and serologic testing, radiologic testing, and pathology, if indicated.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print