Introduction
Renal tubular acidosis (RTA) is a group of diseases characterized by the development of hypokalemic metabolic acidosis with a normal anion gap due to a defect in the ability of the renal tubules to either reabsorb bicarbonate or excrete hydrogen ions in response to acidemia [1,2]. Distal tubular acidosis (dRTA), which was first described in 1946 [3], is also referred to as type 1 RTA and is characterized by the inability to secrete hydrogen ions from the distal tubules. The etiology of dRTA is diverse and can be inherited or acquired. Common clinical presentations of dRTA in the pediatric age group include failure to thrive, polyuria, constipation, rickets, and nephrocalcinosis. We report the case of a 12-year-old Lao girl with long-term untreated dRTA, who presented with severe complications of progressive bony deformities and muscle paralysis.
Case report
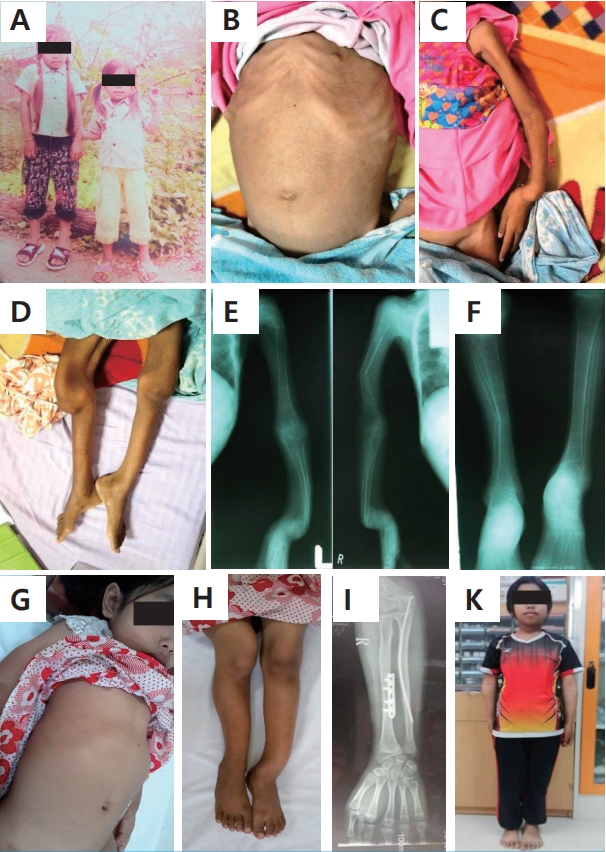
A 12-year-old girl visited the ChildrenŌĆÖs Hospital in Vientiane Capital, Lao PDR with severe bony deformities and progressive paralysis of the extremity muscles. The patient was born via normal vaginal delivery without any perinatal complications. Her birth weight was 2.9 kg. She was breastfed since birth and started solid food at 6 months of age, and she completed vaccination as scheduled. She is the second child of the family, and her growth and development were normal until the age of 8 years. (Fig. 1A) At the age of 8 years, she had a bicycle accident, resulting in dislocation of the right hip and wrist joints. After that, she was bedridden and stayed at home without treatment because of the very challenging socioeconomic status of the family. The range of motion of her upper and lower extremities gradually decreased, accompanied by progressive limb bone deformities. She also lost appetite and weight. At the age of 12 years, the patient visited the outpatient clinic at the ChildrenŌĆÖs Hospital in Vientiane Capital, Lao PDR once but only received a multivitamin prescription without an accurate diagnosis. However, her symptoms gradually worsened resulting in severe contracture of extremity joints and whole-body muscle weakness. Therefore, she visited the ChildrenŌĆÖs Hospital again.
Her father had been healthy until the age of 30 years, when he had progressive paralysis of both arms after working hard, cutting trees. After some treatment and physical therapy at a military hospital, the symptoms of the left arm improved, but the weakness in the right arm continued to this day. Her mother, sister, and cousins were healthy, without any medical condition.
At admission, her body weight was 10 kg (<3rd percentile, she weighed 20 kg at the age of 8 years just before the bicycle accident, according to her father), and her height was 87 cm (<3rd percentile). She was bedridden and looked cachectic. She had a rachitic rosary on the thoracic wall and severe bony deformities with tenderness in the upper and lower extremities. (Fig. 1BŌĆōF) A blood test showed hypokalemia (2.3ŌĆō2.8 mEq/L), metabolic acidosis (serum HCO3ŌĆō15 mEq/L) with normal anion gap (13 mEq/L). Serum calcium level was 8.0 mg/dL, phosphorus 1.0 mmol/L, sodium 135 mEq/L, and chloride 107 mEq/L. Urine pH was high (6.6ŌĆō9.0 by dipstick) and the specific gravity was 1005. There was no proteinuria or hematuria. The urine anion gap was 78 mEq/L. Kidney ultrasonography showed normal sized kidneys with bilateral medullary nephrocalcinosis. Other laboratory tests revealed the following: WBC, 5,500; hematocrit, 38%; hemoglobin, 13 g/dL; platelet, 407,000/mm3; serum creatinine, 0.4 mg/dL; alkaline phosphatase, 1,340 IU/L; and blood sugar, 116 mg/dL. The patient was clinically diagnosed with dRTA and was treated with oral sodium bicarbonate (started from 2.4 g/day and increased to 9.4 g/day), potassium phosphate (4 g/day), vitamin D (400 IU/day), and high calcium diet for 4 months, and then sodium bicarbonate was changed to potassium citrate (Urocitra┬«, Pharmbio Korea, Inc., 3ŌĆō5 mEq/kg/day). Her general condition began to improve after one month of Urocitra treatment, and the follow-up laboratory tests revealed serum HCO3ŌĆō, 21ŌĆō24 mEq/L; potassium, 4.1ŌĆō4.6 mEq/L; and phosphorus, 6ŌĆō6.5 mmol/L. She could stand alone, ride a bike, and attend school after 2 months of treatment. After 6 months of treatment, her body weight and height increased to 15 kg and 98 cm, respectively. After 2.5 years of treatment, she underwent two orthopedic surgeries for correction of the limb bone deformities in the Department of Pediatric Orthopedic Surgery, Seoul National University ChildrenŌĆÖs Hospital, Seoul, Korea with financial support from the Seoul National University ChildrenŌĆÖs Hospital Fund. Two years after the corrective surgery, plate removal from both legs was performed in the Mittaphab Hospital, Lao PDR. (Fig. 1GŌĆōK) Currently, she is 17 years old with a height of 133 cm and taking potassium citrate (Urocitra, 1 mEq/kg/day) only.
Sanger sequencing of the ATP6V1B1, ATP6V0A4, and SLC4A1 genes as well as whole exome sequencing, which was performed in the Department of Pediatrics, Seoul National University ChildrenŌĆÖs Hospital, revealed no pathogenic mutations.
Discussion
The patient had typical biochemical findings of RTA, including metabolic acidosis with a normal anion gap and hypokalemia. Additional findings of alkaline urine, positive urine anion gap, nephrocalcinosis, and rickets suggested that the patient had dRTA. The patient also had growth retardation with severe bony deformities and muscle weakness.
In children, dRTA is almost always observed as a primary entity with underlying genetic causes, and at least three genes are known to cause primary dRTA: ATP6V1B1, ATP6V0A4, and SLC4A1 [4,5]. ATP6V1B1 and ATP6V0A4 encode the B1 and A4 subunits of the apical H+-ATPase pump, that is, the proton pump, in ╬▒-intercalated cells, respectively, and mutations in these genes cause autosomal recessive (AR) forms of dRTA with or without sensorineural hearing loss [6,7]. In a recent large multicenter study by the European dRTA Consortium, the prevalence of hearing loss was 88% in patients with ATP6V1B1 mutations and 36% in patients with ATP6V0A4 mutations [8]. SLC4A1 encodes the basolateral anion (ClŌłÆ/HCO3ŌłÆ) exchanger 1 (AE1) [9]. SLC4A1 mutations can cause dRTA and/or hemolytic anemia with abnormal red cell morphology because AE1 is expressed not only in ╬▒-intercalated cells but also in erythrocytes [9,10]. In non-tropical areas, the majority of SLC4A1 mutations have been reported in association with the autosomal dominant (AD) form of dRTA [9]. However, AR form of dRTA caused by SLC4A1 mutations has been reported mostly from tropical Southeast Asia and other tropical areas, and is therefore called tropical recessive dRTA [11]. We previously reported three Lao patients from two unrelated families with AR dRTA in association with SLC4A1 mutations [12].
GenotypeŌĆōphenotype analysis in the European dRTA Consortium revealed that patients with AD SLC4A1 mutations had a milder phenotype and older onset age compared with those with mutations in the proton pump subunits [8]. While 77% of patients with proton pump subunit mutations presented in the first year of life and all before 10 years of age, 12% of patients with AD SLC4A1 mutations presented in adulthood [8]. However, phenotypes of AR SLC4A1 mutations are quite different from the phenotypes of AD SLC4A1 mutations, but are similar in many respects to the AR dRTA caused by proton pump subunit mutations, such as younger onset age and more severe biochemical changes [11]. The present case showed unusual phenotypes, that is, later disease onset at the age of 8 years (compatible with AD SLC4A1 mutations) with severe biochemical and bone involvement (compatible with the AR form of dRTA). Genetic studies, including whole exome sequencing, in the patient revealed no pathogenic mutations. However, it is difficult to determine whether the patient has a hereditary or acquired form of dRTA because she may have a genetic defect that cannot be detected by Sanger sequencing and whole exome sequencing. In the European dRTA Consortium study [8], 36 of 206 (17.5%) patients who underwent complete genetic tests had no pathogenic mutations. Otherwise, this patient may have a secondary form of dRTA due to prolonged immobilization-induced renal medullary nephrocalcinosis.
Growth failure and/or rickets, resulting from defective bone mineralization in association with chronic metabolic acidosis, are other characteristic features of untreated dRTA patients [13-15]. However, catch-up growth is observed in most patients after the initiation of alkaline treatment [15]. In this patient, bone involvement was unusually severe because she was left in a bed-ridden state for a long time without adequate treatment. The patient also complained of progressive muscle weakness, which may be due to chronic persistent hypokalemia and malnutrition [13]. Biochemical abnormalities and muscle weakness in the patient were normalized by supplementation therapy with alkali, vitamin D, and potassium. However, bony deformities required corrective surgical treatment.
In conclusion, dRTA in children is a controllable disease with favorable long-term prognosis, and accurate diagnosis and proper management are very important. Although this is a single case report of dRTA, the severe phenotypes in this long-term untreated case are of some educational value.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print