Introduction
Hyponatremia and hyperkalemia can develop in various renal and genetic disorders with significant long-term health consequences [1]. Pseudohypoaldosteronism type 1 (PHA1) is one of these disorder characterized by resistance to aldosterone action [2]. It is characterized by renal salt wasting, dehydration, and failure to thrive. The cardinal biochemical features are hyponatremia, hyperkalemia, and metabolic acidosis, despite elevated plasma renin activity and aldosterone levels [3]. There are two different forms of PHA1 that are clinically and genetically distinct, systemic type PHA1 and renal type PHA1. Systemic PHA1 is autosomal recessive inheritance and is characterized by severe resistance to aldosterone action in multiple organs, including the kidney, colon, sweat and salivary glands, and lung. However, renal PHA1 is autosomal dominant inheritance and is characterized by aldosterone resistance restricted to the kidneys [2]. The earliest sign of both type of PHA1 are insufficient weight gain due to chronic dehydration and failure to thrive during infancy [3-5]. Although treatment is often straightforward with oral salt supplementation, the clinical manifestations of this condition can vary widely [6]. A differentiation between systemic and renal PHA1 may be made base on salt requirements, ease of management of electrolyte imbalance, sweat test result and genetic testing.
Here we report a case involving a 28-day-old male infant who only showed poor weight gain at the time of admission and was later diagnosed with renal PHA1 with a novel de novo heterozygous mutation in the NR3C2 gene.
Case report
A 28-day-old male infant was admitted at our hospital because of poor weight gain. There were no other remarkable perinatal complications or previous illness history including urinary tract infection. He was born at term with a body weight of 3.65 kg at birth (50thŌĆō75th percentile). He was the first child of healthy parents, and his family history was unremarkable. At admission, his weight was 3.5 kg (below the 3rd percentile); height, 53.7 cm (25thŌĆō50th percentile), and head circumference, 35.5 cm (10thŌĆō25th percentile). He was in a stable condition with vital signs appropriate for his age (pulse, 140 beats/minute; blood pressure, 82/50 mmHg; respiratory rate, 30/min; and normal peripheral perfusion) and no signs of dehydration. Physical examination revealed no abnormal findings, except for mild scrotal hyperpigmentation. During the 4 weeks of life, the patient consumed adequate amount of milk, and gastrointestinal signs of vomiting and diarrhea were not observed.
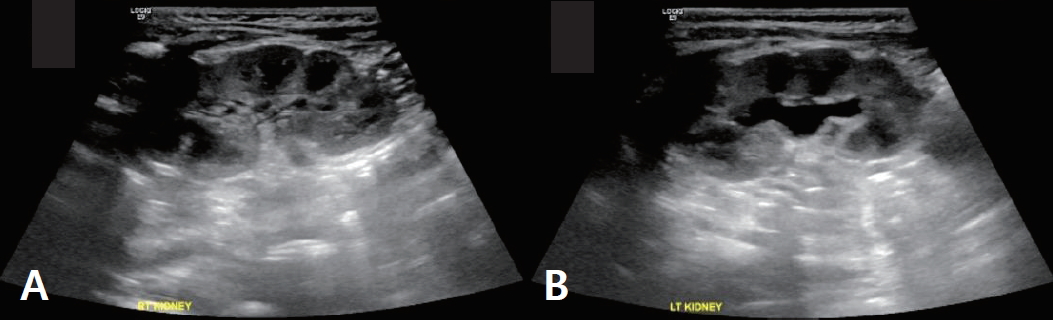
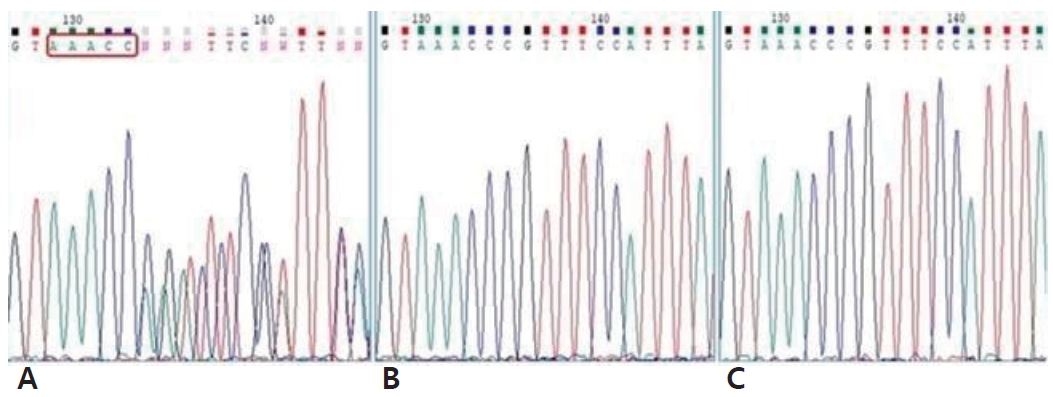
The initial laboratory findings were as follows: serum sodium level, 124 mEq/L; serum potassium level, 7.0 mEq/L; serum chloride level, 97 mEq/L; blood glucose level, 86 mg/dL; blood urea nitrogen level, 21.0 mg/dL; and serum creatinine level, 0.41 mg/dL. The spot urine sodium and potassium levels were 23 and 49.1 mEq/L, respectively. The serum and urine osmolality values were 265 and 267 mOs m/kg, respectively. His venous blood gas analysis revealed a pH of 7.382, pCO2 of 34.8 mmHg, HCO3ŌĆō of 20.2 mmol/L, and base excess of -3.7 mmol/L, reflecting mild metabolic acidosis compensated by respiratory alkalosis. His urinalysis revealed a specific gravity of 1.015, a pH of 5.0, and negative for white blood cells or red blood cells. Renal ultrasonography revealed no abnormality except mild hydronephrosis in left kidneys (Fig. 1). The endocrinological findings at the time of admission were as follows: adrenocorticotropic hormone (ACTH) level, 150.2 pg/mL; cortisol 20.0 ╬╝g/dL, 17-hydroxyprogesterone level, 1.38 ng/mL; DHEA sulfate level 167.4 ╬╝g/dL. However, the ACTH level returned to normal (45 pg/mL) at follow-up. The plasma renin activity and serum aldosterone levels were markedly elevated to 11.45 ng/ml/hr (normal range, 0.32ŌĆō1.84 ng/ml/hr) and 89,955 ng/dL (normal range, 186.5ŌĆō1340 pg/mL), respectively. These results suggested the diagnosis of PHA1; therefore, the Sanger sequencing of the NR3C2 gene was performed. A novel and de novo heterozygous frameshift mutation (c.1341_1345dupAAACC: p.Pro(CCG)449Gln(CAA)Fs*27; Fig. 2) in exon 2 of the NR3C2 gene was detected. Based on the American College of Medical Genetics and Genomics/Association for Molecular Pathology guidelines, this novel variant can be classified as ŌĆ£pathogenicŌĆØ. Direct sequencing for the NR3C2 gene was performed in the parents, however, they displayed the wild type sequence.
The serum electrolyte levels of the patient were normalized with initial parenteral hydration and subsequent oral sodium replacement (7.5 mEq/kg/day). He showed gradual weight gain and is currently 10 months old with height and weight profiles within the 25thŌĆō50th percentile, achieving adequate developmental milestones.
Discussion
Our patient presented with hyponatremia, hyperkalemia, and metabolic acidosis. Differential diagnosis based on the serum electrolyte levels included hypoaldosteronism, adrenal hypoplasia, and congenital adrenal hyperplasia (CAH). We suspected CAH, which is the most frequent cause of electrolyte disturbance, supported by a high initial level of ACTH. However, the ACTH level was decreased to within the normal range at follow up and plasma renin activity and serum aldosterone levels were high, suggesting PHA1. Although we could not precisely account for the initially elevated ACTH level, it is likely that the levels were falsely increased. The ACTH level in blood may be unstable in blood at room temperature; therefore, the methods of blood collection and preparation and storing of plasma may have markedly affected the measured ACTH concentration [7]. If the values are not suitable for the suspected diagnosis, a repeat assessment may be required for confirmation.
PHA1 is a rare syndrome that shows resistance to mineralocorticoid action. PHA1 manifests as two different forms with different levels of severity and phenotypes as follows: systemic type with an autosomal recessive inheritance and renal type with an autosomal dominant inheritance [8]. Systemic PHA1 is caused by mutation in the SCNN1A, SCNN1B, or SCNN1G genes [9], which encode the ╬▒, ╬▓, and ╬│ subunits of the epithelial sodium channel (ENaC) [10]. As ENaC is expressed not only in the distal tubules, but also in the sweat glands, salivary glands, colon, and lung, excessive salt wasting occurs from these organs [11]. Moreover, the salt loss in systemic PHA1 is severe, and symptoms do not improve with age; most patients require lifelong salt supplementation [12]. Renal PHA1 is caused by inactivating mutations of the NR3C2 gene, which encode the mineralocorticoid receptor that is expressed predominantly in the kidney [10], and characterized by aldosterone resistance only in the kidney. The inheritance is mainly autosomal dominant; however, in some sporadic cases, de novo mutations are noted. The main clinical manifestation is insufficient weight gain due to chronic dehydration [4] and biochemical abnormalities, including metabolic acidosis, hyponatremia, and hyperkalemia, with elevated plasma renin activity and aldosterone levels [3]. When compared with systemic PHA1, the symptoms in renal PHA1 are less severe and improve with age in most cases [6,13]. Although mild in nature, renal PHA1 is associated with a high mortality rate among infant [14,15]. Several cases of unexplained neonatal mortality were identified among infants at risk of renal PHA1, which suggests that the seemingly benign renal PHA1 is potentially fatal to neonates [15]. Therefore, early diagnosis and immediate electrolyte correction for neonates at risk of renal PHA1 is important.
In most cases, the symptoms of PHA1 in infants, such as vomiting, poor weight gain, dehydration, and failure to thrive, are non-specific. In addition, the laboratory findings of PHA1 may be confused with other salt-wasting disorders, such as CAH or hypoaldosteronism [2]. For differential diagnosis, the samples should be tested for plasma renin activity, aldosterone, 17-hydroxyprogesterone, ACTH and cortisol levels before the initiation of treatment. In PHA1, metabolic acidosis and hyponatremia that do not improve despite the administration of high-dose mineralocorticoid therapy, increased urine sodium excretion, and hyperkalemia with decreased urine potassium excretion are presented, even if the plasma renin activity and aldosterone levels are high [5]. Our patient presented non-specific symptoms of insufficient weight gain without the characteristics of dehydration or other typical clinical findings at the time of admission. Owing to the rarity of the disease, diagnosis of PHA1 is difficult in the clinical setting. Thus, careful examination, including analysis of electrolyte levels, urinalysis, and hormonal evaluation, if required, should be performed.
In conclusion, we identified a novel mutation in NR3C2 in infant with only poor weight gain. Clinicians should consider PHA1 during differential diagnosis of faltering growth of the common health concerns of the infants.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print