Introduction
Henoch-Sch├Čnlein purpura (HSP) is a form of generalized vasculitis involving the small vessels of the skin, gastrointestinal tract and the kidneys. The exact pathogenesis of HSP remains unknown but is known to develop mostly after the upper respiratory infection. More than 90 percent of patients occur before the age of 10 years. Overall prognosis is good in most patients and no specific treatment is needed. However, some patients progress to end-stage renal disease (ESRD) and the severity of renal symptom is considered to contribute in the long-term outcome of HSP [1]. Abdominal pain is the most common gastrointestinal manifestation and may result from vasculitis, intestinal edema, or complications such as intussusception and perforation. Intussusception, present in about 1.3 to 13.6% of children with HSP, is the most common cause of surgery [2]. Herein we report a case of HSP with intussusception and overt nephritis progressed to end stage renal disease in 15 years of follow-up.
Case report
A 14-year-old boy suffering from skin rash, left ankle arthralgia, and abdominal pain for 6 days visited our pediatric clinic. He had been healthy except a surgery performed 13 years ago for recurrent intussusception. His general condition was not poor on presentation. Vital signs were measured as: blood pressure, 110/60 mmHg; pulse rate, 88 beats/min; and respiration rate, 40/min. He had multiple palpable purpura on lower extremities and painful swelling on left ankle, and distended abdomen with mild tenderness. The laboratory findings were: white blood cell count of 14,800/uL; hemoglobin of 13.0 g/dL; platelet count of 367,000/uL; serum total protein of 6.3 g/dL; albumin of 3.4 g/dL; serum creatinine of 0.6 mg/dL; serum C3 of 56 mg/dL; C4 of 23 mg/dL; and antistreptolysin-O (ASO) of 97 IU/mL. Urinalysis showed no blood or protein. The chest and abdominal x-rays were normal.
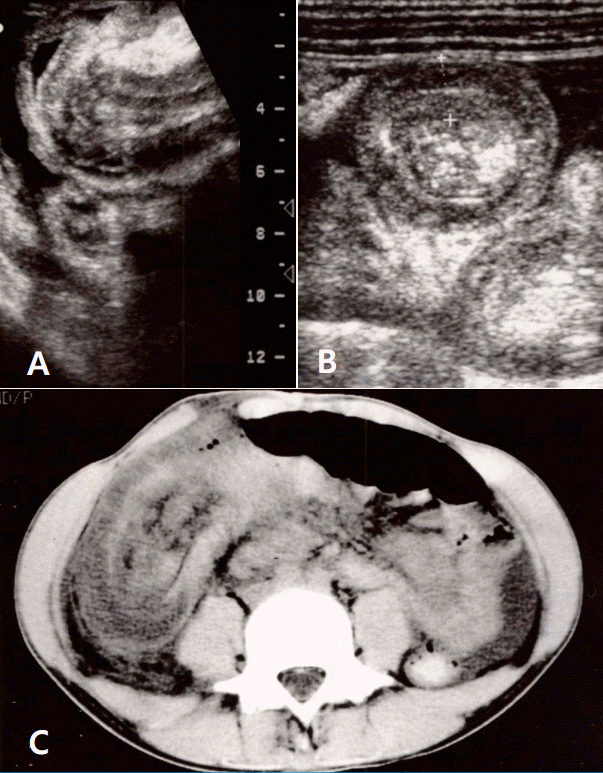
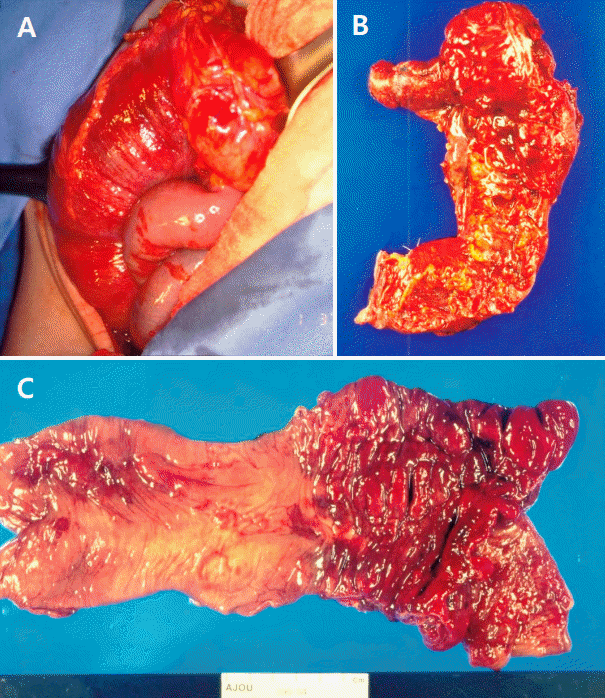
The patient was hospitalized with a diagnosis of HSP involving skin, gastrointestinal system and joints. Intravenous dexamethasone (0.1 mg/kg per dose every 6 hours) was prescribed from the first day of admission (hospital day #1, HD #1), and the patient was stabilized for the next few days. On HD #5, he developed a sudden clamping abdominal pain with vomiting, bloody loose stool and dark brown urine. Ultrasonography (US) revealed a multilayered mass in the bowel lumen along the periumbilical area and contrast-enhanced computed tomography demonstrated the classic bowel-in-bowel configuration (Fig. 1). The diagnosis of intussusception was established and an emergent laparotomy was performed. At laparotomy, ileocolocolic-type intussusception was found. The entire small bowel loops were edematous, and multiple submucosal hemorrhages were noted. Manual reduction failed to revive the involved segments, thus, right hemicolectomy with ileotransverse colostomy was performed (Fig. 2).
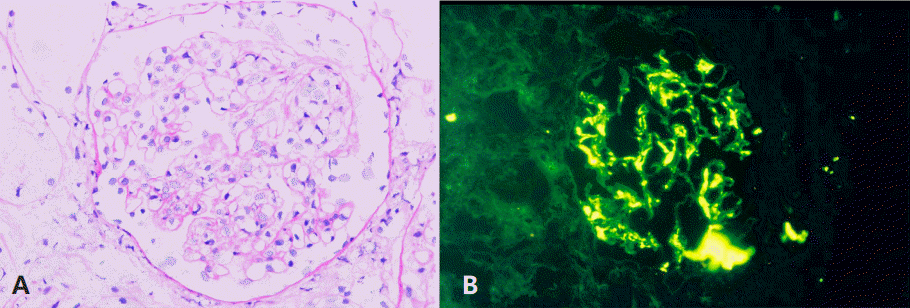
Urine test on HD #7 showed: specific gravity (SG) of 1.030; 3+ blood; 2+ protein; and many red blood cells (RBCs) per high-power field. Urinary protein excretion increased from 2,351 mg/24hr on HD #12 to 25,992 mg/24 hr on HD #29. Despite 4 weeks of steroid therapy, the illness evolved to overt nephrotic syndrome with hypertension. Angiotensin converting enzyme (ACE) inhibitor and furosemide were prescribed. Renal biopsy was performed on HD# 43 and showed diffuse mesangial proliferation with occasional segmental sclerosis. Small cellular or fibrocellular crescents were noted in 5 out of 13 glomeruli (ISKDC class IIIa). Tubular atrophy or interstitial fibrosis was not present. Immunofluorescence for IgA showed strong staining at mesangium (Fig. 3).
The patient was under successful control for the intermittent abdominal pain and vomiting until HD #55, but on HD #56, after 3 days of uncontrolled hypertension (>180/120 mmHg), suddenly started a generalized tonic-clonic seizure. The first convulsion responded to intravenous diazepam, but there were two additional seizures in the following 24 hours. Brain CT scan demonstrated no specific findings other than mild dilatation of both ventricles. The electroencephalogram (EEG) findings were normal and there were no more seizures. Urinalysis on HD #80 showed 3+ blood and 3+ protein, but the amount of protein excretion decreased to 5,332 mg/24hr. Cyclosporine A (CsA) therapy (2 mg/kg/day PO divided into 2 doses) was started on HD #80 and steroid tapering was done within the next 30 days. The patient was in good condition and discharged on HD #90.
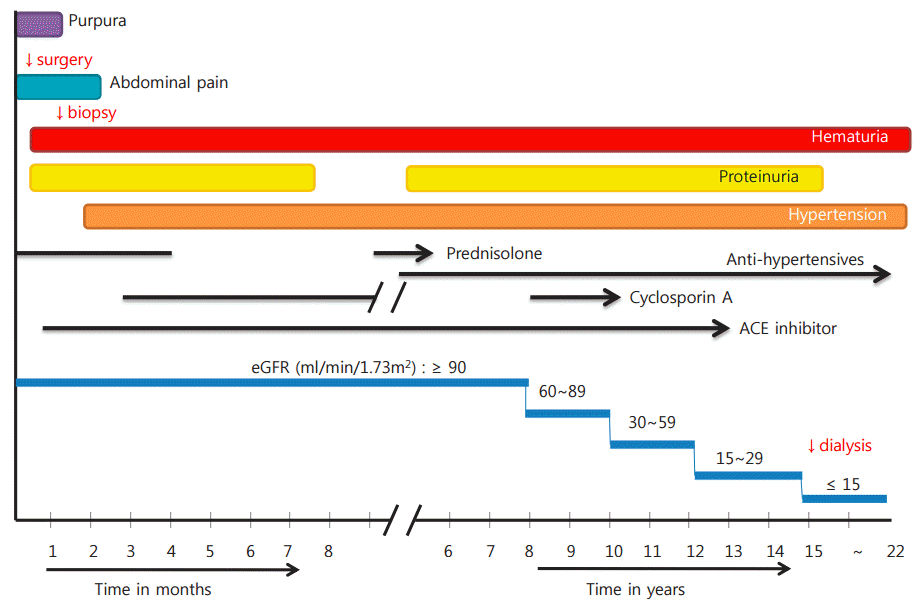
After discharge, he had been regularly followed up for blood and urine tests to check kidney function, including CsA levels. Microscopic hematuria continued but remission of proteinuria was achieved 5 months after the CsA therapy. After 12 months of CsA regimens, the patient was transferred to another hospital where he was treated for HSP nephritis recurrences with CsA regimens. Seven years after the onset of HSP, the patient visited our hospital again with persistent proteinuria/hematuria and hypertension, and was prescribed medications such as diuretics, angiotensin receptor blocker, statins, and calcium and vitamin D supplements. No further visits were made again by the patient for 1 year. He then revisited with sudden onset of severe headache and hypertension of 200/120 mmHg. On assessments, serum electrolytes, blood glucose and calcium concentrations were within normal limits. Urine dipstick was 1+ blood and 3+ protein. The estimated glomerular filtration rate (eGFR) of the patient reached 59 ml/min/1.73m2 (chronic kidney disease stage 3) after 10 years, and <15 ml/min/1.73m2 (ESRD) after 15 years. He is now on hemodialysis and waiting for renal transplant (Fig. 4).
Discussion
HSP is a systemic vasculitis disorder first reported by Heberden in 1806. Sch├Čnlein described the association of purpura and joint pain, and he termed it ŌĆśpeliosis rheumaticaŌĆÖ in 1837. Henoch added gastrointestinal and renal manifestations in 1874 and 1899, respectively [3].
Although various infectious insults may be associated with development of HSP, the pathogenesis of HSP and its complications such as intussusception and renal injury is still unknown. However, it is believed that the substances which induce inflammation in various pathologic lesions of HSP, were released. These substances may have affinity to target host calls such as skin vascular cells (purpura), intestinal vascular cells (abdominal pain), joint cells (arthritis), renal cells, and rarely other organ cells. However, the substances may be originated from not only pathogens but also the host cells injured by various insults. It was previously proposed that immune cells and immune proteins may control these substances toxic to host cells, depended upon the size and biochemical characteristics of the substances. Thus, the substance and corresponding immune cells may be responsible for inducing tissue inflammation and repair in HSP. Although the immunopathogenesis of HSP, from skin purpura to severe nephritis may be complex, the host immune/repair system controls the disease progress and decides prognosis of the disease. The prognosis of HSP nephritis may be dependent on whether the host immune/repair system could control etiological substances and other toxic substances or not [4].
The incidence of gastrointestinal manifestations of HSP is about 50-75% and most of them are treated conservatively. Abdominal pain may be due to hemorrhage and edema within intestinal wall or life-threatening complications such as perforation, obstruction, infarction, hemorrhage and intussusceptions [5-7]. Intussusception is the most common surgical complication of HSP. Unlike typical intussusceptions, which are ileo-colic and can be diagnosed and reduced by barium enema, the sites of intussusception in HSP are most frequently ileo-ileal (51.4%), followed by ileocolic (38.6%) and jejuno-jejunal (7%). HSP associated intussusception generally occurs in children >2 years of age. It is very difficult to distinguish HSP complicating intussusception from inflammatory abdominal pain found in simple HSP [8,9]. In our case, we diagnosed intussusception with ultrasonography and confirmed it as ileocolocolic-type by laparotomy.
Renal involvement has been reported to occur in 20-50 % of children with HSP. Among these patients with HSP nephritis, about 5% progress to ESRD at 5 years [10,11]. The clinical spectrum of renal manifestations varies from isolated microscopic hematuria to nephritic/nephrotic syndrome with renal failure. According to reports, the risk of ESRD in HSP nephritis is related to the severity of renal manifestations; less than 5% in patients with hematuria and/or minimal proteinuria, 15% in those with heavy proteinuria, and over 50% in those with nephritic and/or nephrotic syndromes [12]. Although renal involvement occurs within 3 months of the onset of HSP in most patients, follow-up should be done for at least 6 months, including urine test and blood pressure measurement. In our case, renal involvement began suddenly 10 days after the onset of HSP concomitant with intussusception, and progressed rapidly to a nephritic-nephrotic nature.
Treatment for HSP remains primarily supportive in most cases of HSP, due to its self-limited symptoms over time. Non-steroidal anti-inflammatory drugs (NSAIDs) may help joint pain, and steroids may reduce abdominal pain. Children with severe HSP nephritis have been treated with steroid pulse therapy or in combination with immunosuppressants such as azathioprine or cyclosporine A to prevent renal damages. But the management of HSP nephritis still remains highly controversial [13].
After one year of successful initial cyclosporine A therapy, nephritis and nephrosis recurred, showed no response to CsA again, and progressed to ESRD in 15 years, in our case. Still, there is no consensus on the optimal treatment for HSP nephritis. Our suggestion is that immunotherapy shall be prolonged and initiated more promptly to patients with fulminant complications, such as overt nephritis and intussusception.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print