Introduction
Gorham-Stout Syndrome (GSS) is one of the rare syndromes, also known as vanishing bone or phantom bone disease, characterized by spontaneous and progressive osteolysis. Worldwide, only 200 affected individuals have been reported [1]. Common symptoms include bone pain, swelling, osseous deformity, and pathologic fractures of the extremities. In addition to progressive bone loss commonly involving the clavicles, ribs, and spine, it is also characterized by proliferation of vascular and lymphatic channels in the involved bony segments. It is recognized as an aggressive form of skeletal angiomatosis disease [2]. The proliferation of lymphatic vessels may result in chylothorax and pleural effusion. Hardegger et al. [3] classified ŌĆ£idiopathic osteolysisŌĆØ into five classes as follows: type 1, hereditary multicentric carpotarsal osteolysis syndrome (MCTO, OMIM #166300) with dominant transmission from MAFB gene mutation [4]; type 2, hereditary multicentric osteolysis with recessive transmission, which is consistent with type 1 in addition to generalized osteoporosis; type 3, nonhereditary multicentric osteolysis with nephropathy; type 4, GSS; and type 5, Winchester syndrome (OMIM #277950), defined as a monocentric disease of autosomal recessive transmission. Of these, kidney involvement has been reported in type 1 and 3, and a causative gene was identified only for type 1. For type 4 idiopathic osteolysis, GSS, no genetic cause was identified and the etiology remains unclear. Treatment of GSS is largely symptomatic, including surgical treatment, radiotherapy and medications of bisphosphonates, vitamin D, interferon alfa and sirolimus as a novel therapy [1]. Herein we report a case of focal segmental glomerulosclerosis (FSGS) diagnosed in a patient with GSS.
Case report
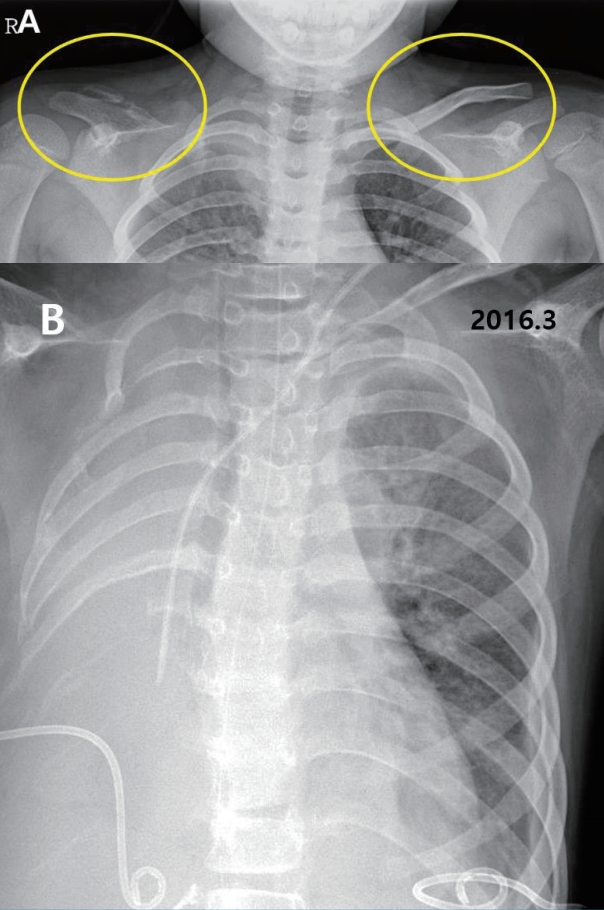
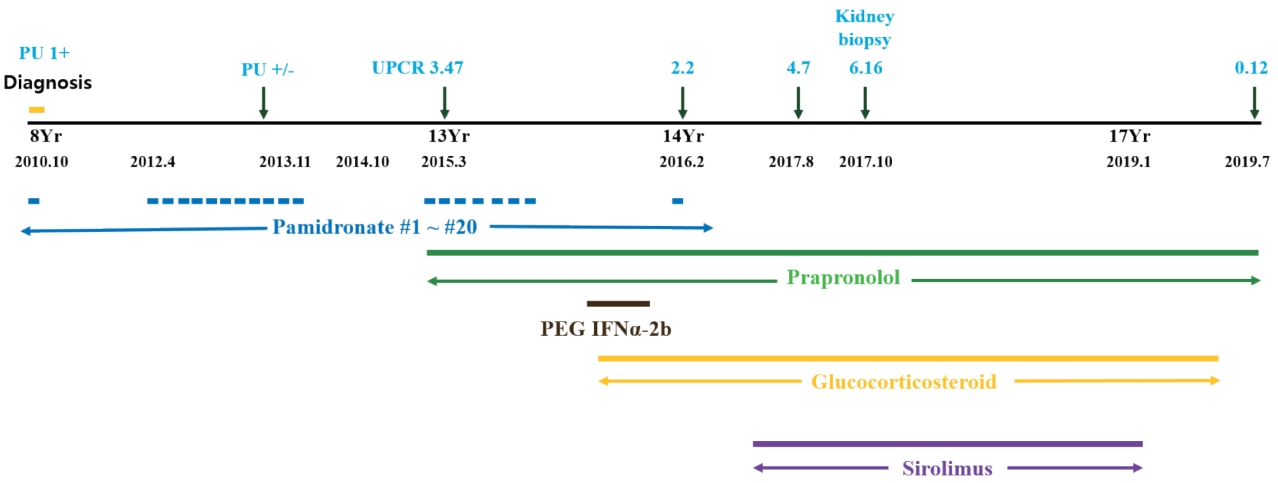
A 15-year-old boy with GSS was referred to the Division of Pediatric Nephrology for proteinuria in August 2017. He had recurrent clavicular fractures since the age of 4 and was diagnosed with GSS in 2010, at age 8, based on clinical findings of pleural effusion, recurrent fracture and radiological finding of clavicle osteolysis (Fig. 1). Multiple destructive lesions were noted in the clavicles, multiple ribs, and spine. Pamidronate was administered a total of 20 times either every 1ŌĆō2 months (2ndŌĆō19th dose) or intermittently (0.5ŌĆō1.5 year interval) from Oct 2010 to Feb 2016 (Fig. 2) to decrease osteolysis. Pleural effusions recurred intermittently, beginning in February 2015, requiring hospitalization for chest pain and dyspnea. Multiple therapies were administered in an attempt to alleviate the recurrent pleural effusions. Propranolol was administered beginning in March 2015 to decrease VEGF-A levels, interferon alfa was administered from October 2015 to February 2016 for its anti-angiogenic effect and immunity enhancement, and glucocorticosteroids starting in November 2015 and sirolimus beginning in May 2016 to suppress lymphatic proliferation. Although he had experienced respiratory failure several times requiring ventilator care and eventually a tracheostomy, the disease has been under control with these medications with the addition of sirolimus (Fig. 2). Proteinuria was first noted in April 2015, coinciding with an infection and intermittently noted , but was not investigated further until April 2016. Proteinuria was again noted when he was hospitalized for pneumonia in Aug 2017.
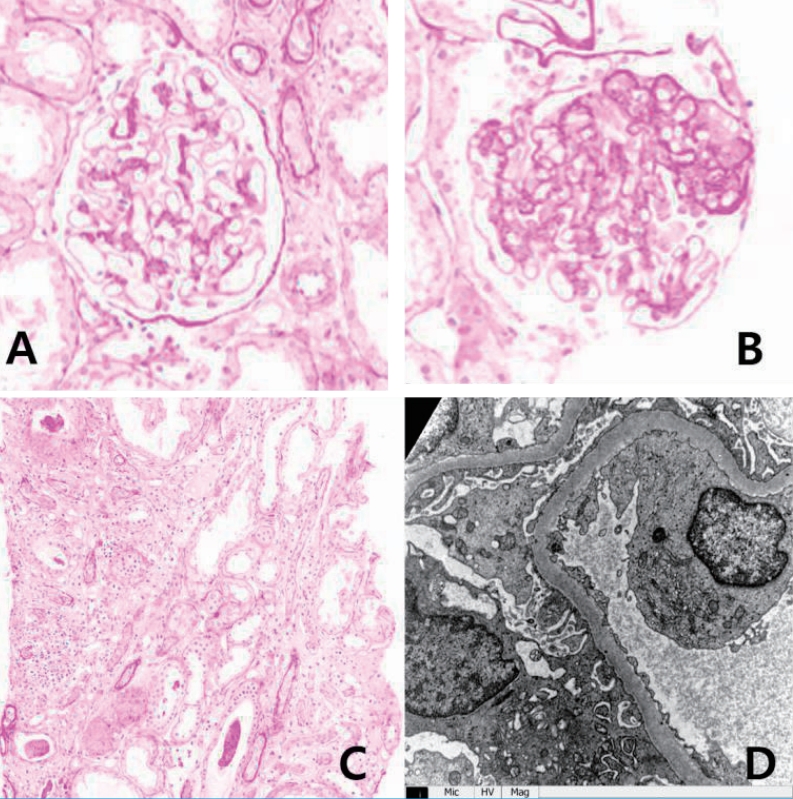
On physical examination in Aug 2017, his height was 150 cm (<3 percentile) and weight was 50.1 kg (10ŌĆō25 percentile). He had normal growth until the age of 12 (height, 25ŌĆō50 percentile; weight, 50ŌĆō75 percentile), but experienced growth restriction thereafter. His blood pressure was 124/75 mmHg (both 75ŌĆō90 percentile), pulse 143 beats/min, respiratory rate 28 breaths/min, body temperature 38.7Ōäā and SpO2 92% on room air. Breath sounds were coarse with rales in both lung fields and decreased due to chronic pleural effusion. He urinated well and was not edematous and there was no pitting edema. Laboratory tests revealed hypoalbuminemia (3.0 g/dL; normal, 3.3ŌĆō5.2 g/dL) and proteinuria (urine protein to creatinine ratio 4.7ŌĆō10.2; normal, <0.2) (Fig. 2). Serum total protein was low (5.8 g/dL; normal, 6.0ŌĆō8.0 g/dL), serum electrolytes, blood urea nitrogen, creatinine, total cholesterol, and liver enzymes were all within normal limits. Urinalysis showed intermittent mild microscopic hematuria (5ŌĆō9 RBC/HPF). Urine beta 2-microglobulin and N-acetyl-beta-D-glucosaminidase (NAG)/creatinine ratio were increased to 4.32 (0ŌĆō0.37 ╬╝g/mL) and 34.2 (0ŌĆō5.6 IU/g*Cr), respectively. Renal ultrasound showed diffusely increased renal parenchymal echogenicity with a normal kidney size (Fig. 3). Renal biopsy was performed in October 2017 and revealed FSGS, not otherwise specified (NOS). Microscopic examination found 1 of 14 (7.1%) glomeruli with segmental sclerosis and focal mild hypercellularity involving mesangial cells. Focal mild tubular atrophy and injury, focal slight fibrosis, and mild interstitial inflammatory cell (mononuclear cells and eosinophils) infiltration was also noted. Immunofluorescence and electron microscopic findings were unremarkable (Fig. 3). Electron microscopic findings showed a normal glomerular basement membrane with mild focal effacement of foot processes. There were no electron-dense deposits.
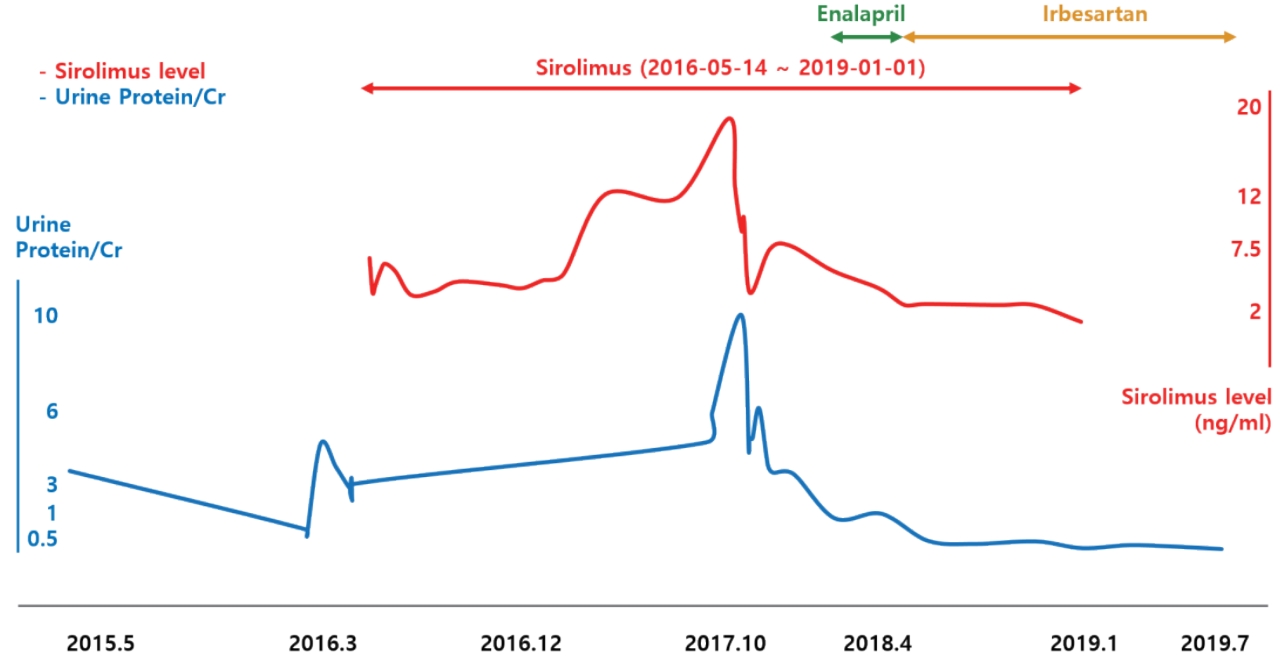
Given the possibilities of GSS-associated or drug induced glomerulopathy, in order to decrease proteinuria, enalapril, an angiotensin converting enzyme inhibitor was started in March 2017 and was changed to irbesartan, an angiotensin II receptor antagonist, in December 2017. Additionally, sirolimus was tapered over concerns of sirolimus-induced proteinuria. His proteinuria decreased gradually after starting enalapril and reducing the sirolimus, and once irbesartan was initiated, the proteinuria decreased to less than nephrotic range (urine protein to creatinine ratio of 1.64 in April 2018). Serum albumin has remained in the normal range (>3.6 g/dL) since November 2017. His GSS is currently well controlled with no more pleural effusions despite the reduction of sirolimus. In January 2019 sirolimus was discontinued. Since then, proteinuria has decreased further and finally disappeared in July 2019. Currently, the disease is well controlled without proteinuria on propranolol and irbesartan.
Discussion
This case report presents a patient with GSS complicated by FSGS with proteinuria. To our knowledge, there are no reports of an association between GSS and glomerular disease leading to the question of the pathogenesis of FSGS in this patient. Is it an unknown manifestation of GSS itself, or is it secondary to his medications? In fact, pamidronate, sirolimus, and interferon alpha are well known to cause proteinuria and FSGS.
Pamidronate, a bisphosphonate drug, can cause glomerular diseases, such as a collapsing variant of FSGS, or less frequently FSGS NOS or minimal change disease [5]. It has been linked to proteinuria that usually begins 3 to 42 months after the initiation of therapy [6]. Remission of the glomerular disease following withdrawal of pamidronate persists in many patients; however, some have progressed to end-stage kidney disease despite cessation of the drug [7]. Barri et al. [5] suggested that more frequent pamidronate infusion (total number varied from 10 to 40 or more) could be correlated with the incidence of proteinuria and FSGS. In GSS, 6 to over 10 times infusions had been reported [8,9]. Our patient received pamidronate a total of 20 times given either every 1ŌĆō2 months or at a 0.5ŌĆō1.5 year interval (Fig. 2) and he developed proteinuria 3 years after the initiation of this frequent pamidronate treatment. Additionally, his proteinuria persisted for more than one and a half years following withdrawal of pamidronate. Therefore, it is possible that his proteinuria was initiated by the pamidronate.
The second candidate drug is sirolimus, which is used to treat nephrotic syndrome or proteinuria, but it is known to induce proteinuria in a dose-dependent manner as well. Letavernier et al. [10] reported that up to 64% of patients with chronic allograft nephropathy had massive proteinuria after switching their medication from calcineurin inhibitors (CNI) to sirolimus, and 30% of them developed FSGS. Moreover, de novo sirolimus glomerular toxicity has also been described in patients who never received CNI [11]. Besides kidney transplantation, Cho et al. [12] reported that sirolimus might exacerbate preexisting idiopathic FSGS resulting in worse proteinuria. Our patientŌĆÖs proteinuria increased into the nephrotic range after the addition of sirolimus and improved in a dose-dependent manner with its reduction (Fig. 4). It is highly likely that sirolimus was associated with the worsening of his proteinuria, whether it caused FSGS or exacerbated preexisting FSGS.
Although interferon alpha is also known to cause FSGS [13], it was used only for short time in our patient. Propranolol, another medication used by our patient, has not been reported to cause proteinuria or FSGS. Therefore it is unlikely that his FSGS was caused by either of these two drugs.
Renal dysfunction with osteolysis syndrome syndrome is only known to occur in type 1 (MCTO) and type 3 osteolysis. MCTO from mutation of the MAFB features robust osteoclastogenesis, especially at the carpal and tarsal bones [14]. The MAFB gene, expressed in glomerular podocytes as well as osteoclasts, plays a crucial role in renal development, and its mutation has been associated with FSGS [15]. Destructive lesions in the hands and feet which are the characteristics type 1 or 3 osteolysis were not evident in this case. And kidney involvement has not been reported in GSS. Therefore, it is reasonable to assume that this proteinuria was caused by secondary etiology such as medications. In addition, increased urine beta 2-microglobulin and NAG/creatinine ratio could be considered to suggest kidney tubular injury, probably caused by certain drugs.
The sequential changes of proteinuria in association with medication changes suggest that the pathogenesis of the proteinuria may be as follows: the proteinuria was initially caused by pamidronate and decreased (or disappeared) following discontinuation of the drug. Sirolimus either aggravated or reinduced proteinuria and FSGS.
In conclusion, we report a rare case of GSS presenting with proteinuria and FSGS which are unexpected findings in GSS. Since FSGS-inducing medications, pamidronate and sirolimus, are indicated for the treatment of osteolysis, it is easy to overlook, but when these medications are used, it is important to monitor the development of proteinuria in this very rare type of vanishing bone syndrome. Nevertheless, we cannot rule out the possibility that this FSGS might be a new findings for GSS.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print